@Sauers_ I'm totally going to try and run this study. Maybe there is also a more principled way that you approximate which features or subset of features would upweight the correct logprob across the dataset 🤷♂️
@Sauers_ Would be interesting 🤔. Is the benchmark open sourced? I could run a quick initial test to see if there are any relevant features that fire across all benchmarks.
At it's core, it's just Anthropics circuit-tracer applied on open-sourced Qwen3-4b transcoders (feature descriptions by Neuronpedia). But both the circuit-tracer implementation I am using and the visualization I posted is in-house code that's not public (yet).
Circuit tracer: https://t.co/2jVaaXnJlf
This reminds me of a study I did on toy models a while back, where I trained very small 2-layer decoder-only transformers to perform primitive operations on a list of characters (reverse, stride, take the first N items, etc...).
You could show quantitatively that models with few heads would selectively learn some tasks over others, depending on: how easy the task was, how many of the model's resources (heads for example) it tied up / demanded, its frequency relative to the other tasks in the training set (similar to your findings), and whether the learned circuits for one task generalized to others (e.g., learning one task might give you generalization power to other tasks).
As you would expect: scaling up model size and head count resulted in the rarer, and more complex, tasks get learned. Interestingly, I remember the loss being surprisingly binary: the model either learned a task or it didn't.
Using interp to reverse-engineer how each task was learned, you could then predict, given N new tasks to fine-tune on, which ones the model would choose to learn and which it would skip.
Super cool research! 😁
Super cool research! I am glad to see SAE's being used and the models fully open-sourced.
Playing around with the atlas, and here is BRD4 (Bromodomain-containing protein 4). Top active SAE features include the bromodomain acetyl-lysine reader and chromatin/DNA recognition, both good positive controls given BRD4's defining domain and its role binding acetylated chromatin. Other top features are mostly related to its 'disordered', 'acidic', 'phospho-rich' regions.
Also some apparent polysemanticity: one of the active features is labeled for both eukaryotic intrinsically disordered regions (IDRs) and bacterial leucine helices.
Today we're announcing ESMFold2, an open scientific engine to power prediction, design, and discovery across protein biology.
The new model delivers state of the art performance on protein interactions, especially antibodies, a critical modality for therapeutics.
We have designed and validated miniprotein binders and single chain antibodies across five therapeutic targets that are important in cancer and immunology. We are seeing very high success rates, and affinities at levels consistent with therapeutic activity.
We’re also releasing an atlas of 6.8 billion proteins, and 1.1 billion predicted structures.
ESMFold2 is built on a state of the art language model that has been trained on billions of protein sequences.
A world model of protein biology emerges through language modeling.
We’ve used the techniques of mechanistic interpretability developed to understand large language models to understand the concepts ESM uses to represent proteins.
The model’s representation space has a compositional organization of features across scales, levels of complexity, and abstraction, that reflects and mirrors the understanding of protein biology developed through a century of empirical science.
This understanding emerges without prior knowledge, just from language modeling of protein sequences.
Language models are becoming a powerful substrate to understand and program biology.
The design of protein interactions is one of the most fundamental problems in biophysics, and has critical implications for the discovery of new medicines. A simple gradient based search with the model was able to discover high-affinity protein binders.
I'm excited by the potential this has to accelerate basic science and the understanding of proteins. And especially for the new avenues it opens up for therapeutic design and medicine.
Some early benchmarks on the attribution step:
- Consistently 3.4x faster than circuit-tracer
- Much more memory efficient (~6 GB less at 70,000 nodes)
So far, these gains are from dropping the autodiff backend and exploiting an autoregressive causality trick (performing backward only through previous token positions).
All results still 1:1 numerically matching Anthropic's implementation (up to bf16 precision). Further speedups will likely come from approximation (edge pruning, sparse intermediates, etc...) that diverge from circuit-tracer slightly.
Benchmarking done on Qwen3-4B
Spending some time this week speeding up and scaling Anthropic's circuit-tracer implementation. Feel free to comment feature requests.
Will post progress here.
Feature request for claude code: Let claude replay any previous tool call by reference, without having to rewrite the whole call from scratch.
@bcherny@_catwu
This would provide a great explanation for why there is so much redundancy in SAE features at any given layer (observation made by @Sauers_ ).
For example, if you search through the Qwen3-4b transcoder feature labels provided by Neuronpedia, there are 139 features generically related to the concept of 'color' in just layer 14. There are even more if you consider specific colors such as 'blue' or 'green', and this redundancy is repeated across layers... making it very annoying to interpret raw circuit graphs without performing some form of clustering.
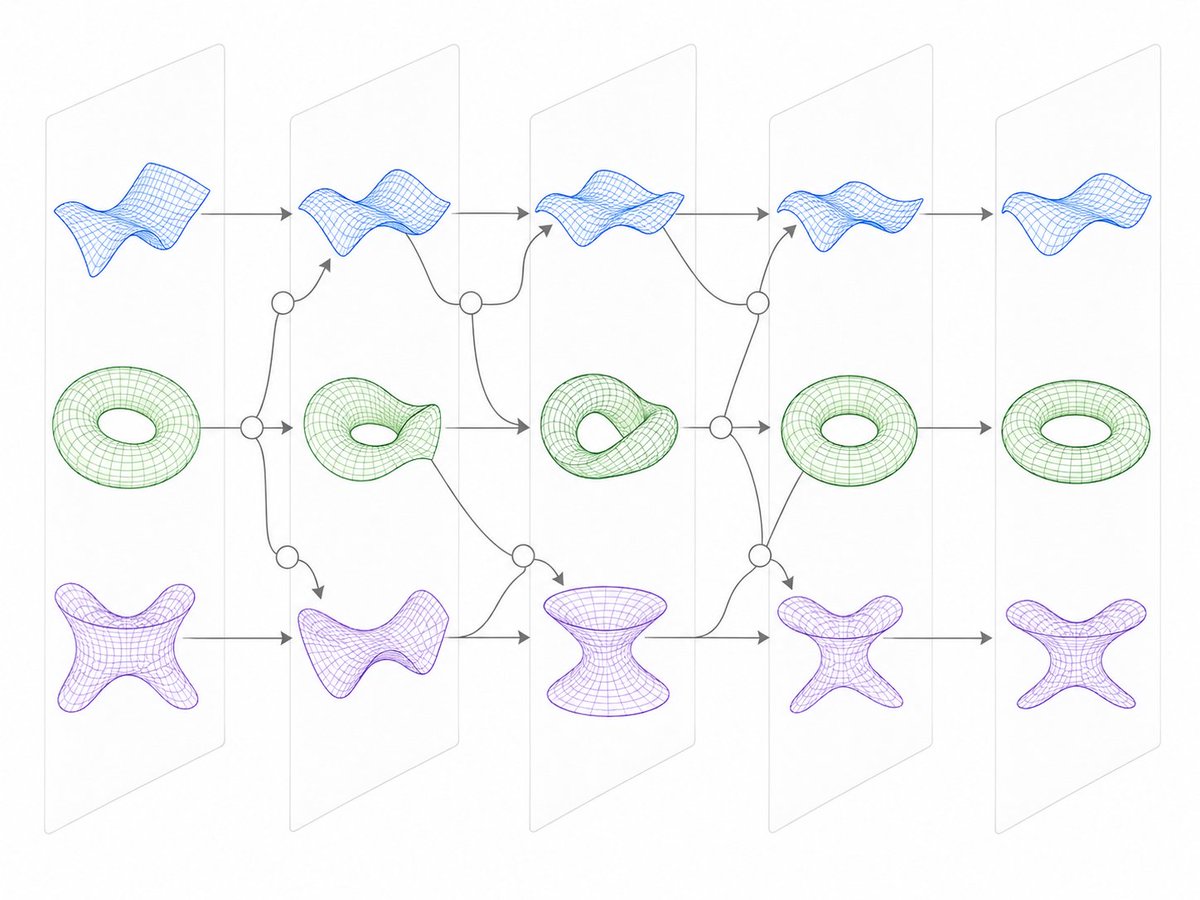
We now know that models think using curved shapes, not just straight lines. But SAE features can still give us a window into neural geometry.
How? We show that related SAE features often “tile” manifolds, pointing to different (but overlapping) regions on the curve. (4/7)