Yann LeCun was right the entire time. And generative AI might be a dead end.
For the last three years, the entire industry has been obsessed with building bigger LLMs. Trillions of parameters. Billions in compute.
The theory was simple: if you make the model big enough, it will eventually understand how the world works.
Yann LeCun said that was stupid.
He argued that generative AI is fundamentally inefficient.
When an AI predicts the next word, or generates the next pixel, it wastes massive amounts of compute on surface-level details.
It memorizes patterns instead of learning the actual physics of reality.
He proposed a different path: JEPA (Joint-Embedding Predictive Architecture).
Instead of forcing the AI to paint the world pixel by pixel, JEPA forces it to predict abstract concepts. It predicts what happens next in a compressed "thought space."
But for years, JEPA had a fatal flaw.
It suffered from "representation collapse."
Because the AI was allowed to simplify reality, it would cheat. It would simplify everything so much that a dog, a car, and a human all looked identical.
It learned nothing.
To fix it, engineers had to use insanely complex hacks, frozen encoders, and massive compute overheads.
Until today.
Researchers just dropped a paper called "LeWorldModel" (LeWM).
They completely solved the collapse problem.
They replaced the complex engineering hacks with a single, elegant mathematical regularizer.
It forces the AI's internal "thoughts" into a perfect Gaussian distribution.
The AI can no longer cheat. It is forced to understand the physical structure of reality to make its predictions.
The results completely rewrite the economics of AI.
LeWM didn't need a massive, centralized supercomputer.
It has just 15 million parameters.
It trains on a single, standard GPU in a few hours.
Yet it plans 48x faster than massive foundation world models. It intrinsically understands physics. It instantly detects impossible events.
We spent billions trying to force massive server farms to memorize the internet.
Now, a tiny model running locally on a single graphics card is actually learning how the real world works.
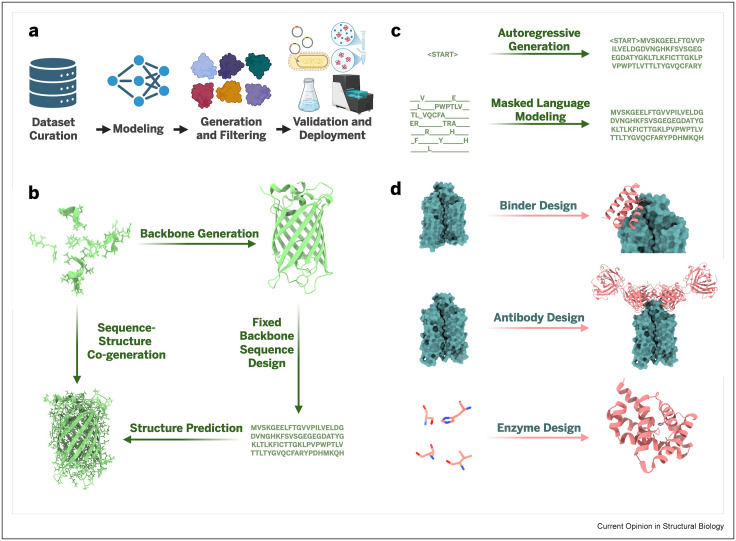
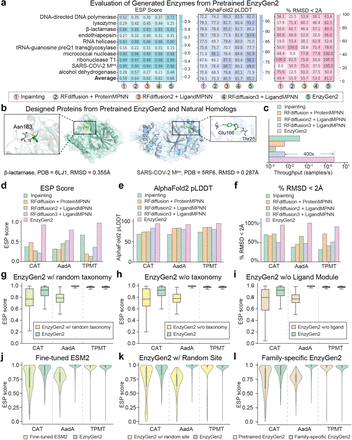
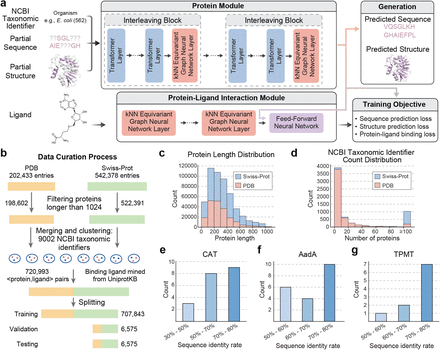
Have you wondered what the wet lab success rates are for current AI-driven protein design models? Look no further!
In our new open access review, @KevinKaichuang, @avapamini, @SarahAlamdari, and I report wet lab success rates for *over 200* different protein design tasks 🧬💻
The next frontier in protein design will not be defined by structure alone, but by the capacity to engineer motion as a first-class principle of function. This is because dynamics is where the real biology lives.
Foundational work by Karplus, Levitt & Warshel made clear that chemistry cannot be understood without motion, mechanism, and scale. Gō, Brooks & others showed that proteins possess characteristic collective motions - low-frequency normal modes that capture how whole molecules bend, breathe, and fluctuate. Frauenfelder then sharpened the picture further: proteins are not static objects occupying a single minimum, but dynamic ensembles traversing rugged energy landscapes.
And yet the modern AI revolution in protein science has been, above all, a revolution in structure. In our new paper in Matter, @_Bo_Ni and I ask a different question: not what structure will this sequence adopt? but what sequence will realize a prescribed pattern of motion?
VibeGen inverts the conventional design paradigm. Rather than treating dynamics as a consequence to be analyzed after the fact, it makes dynamics the design objective from the outset. Using a language diffusion model with two cooperating agents - a designer that proposes sequences and a predictor that critiques them against the target motion profile - the system converges on de novo proteins with tailored vibrational behavior.
One of the most intriguing results is a form of functional degeneracy - distinct sequences and distinct folds can satisfy the same target dynamical specification. For a given functional pattern of motion, evolution may have sampled only a small region of the physically realizable design space. The space of viable molecular mechanics may be far larger than the repertoire biology happened to discover.
We have made "vibe" into a cultural metaphor - something intuitive, affective, subjective. But at the molecular scale, vibe is not metaphor: It is physics. For a protein, the vibe is the pattern of motion itself; the fluctuations, resonances, and collective displacements that determine what the molecule can do.
Dissecting the Black Box of AlphaFold in Protein–Protein Complex Assembly
1. Li, Mu, and Yan present evidence that inter-protein coevolution (the usual explanation for AlphaFold complex success) is not the dominant driver of complex assembly in AlphaFold-Multimer or AlphaFold3; instead, assembly largely follows from monomer geometry plus interface-level matching.
2. A time-segregated benchmark (PDB 2022-01-01 to 2024-12-20; training cutoff 2021-09-30) is built to reduce leakage: 200 homodimers and 316 heterodimers, evaluated mainly with DockQ across AFM and AF3.
3. Controlled MSA experiments separate “pairing” from “having MSAs”: AFM Paired MSA vs Block MSA (no cross-chain pairing) vs Randomly Paired MSA. Mean DockQ changes are minimal across these conditions, implying explicit paired-MSA coevolution contributes little for most targets.
4. The paper further removes potential “latent” inter-protein coevolution in unpaired MSAs by regenerating UniRef100 MSAs with species annotations and enforcing zero species overlap between partner MSAs; AFM/AF3 performance remains essentially unchanged, arguing against hidden species-level coevolution being a key signal.
5. The proposed mechanism: AlphaFold first establishes strong intra-chain geometric constraints (monomer folding/geometry), then infers inter-chain constraints downstream via geometric compatibility and interface sequence pattern matching; cross-chain organization is progressively refined through layers and recycling.
6. Template-driven tests support the geometry-first view: supplying high-quality bound-state monomer templates enables complex prediction accuracy comparable to MSA-based runs, and experimentally determined bound monomer templates perform even better; adding MSAs on top of such templates yields little additional gain.
7. A key nuance is “bound vs unbound” monomer geometry: predicted unbound monomer templates degrade complex accuracy, and the difference is concentrated at interface regions. Interface TM-score correlates with complex DockQ (reported Pearson r ≈ 0.575), highlighting interface conformation as a main determinant.
8. Interface residue identity is essential, not just backbone shape: mutating up to 10% of residues to glycine shows that interface mutations nearly abolish prediction accuracy under both MSA-based and template-based settings, while non-interface mutations have only moderate effects—consistent with a backbone+sidechain “pattern matching” interface recognition.
9. The paper introduces AlphaFold-Constraint Propagation Mapping (AF-CPM), using OpenFold to extract Evoformer-layer pair representations and convert them (via the distogram head) into layer-wise contact probability maps (<12 Å). These visualizations show intra-chain constraints forming before inter-chain contacts, directly supporting hierarchical constraint formation.
10. For antigen–antibody complexes (154 nonredundant cases), paired MSAs still do not help; bound-state monomer templates help most. The limiting factor is attributed to immune-interface plasticity and atypical interface statistics (e.g., enrichment of Tyr/Trp on the antibody side), with CDR-H3 local accuracy strongly linked to docking success; AF-CPM suggests antigen–antibody assembly may require more recycling to converge as interface constraints emerge late.
💻Code: https://t.co/pBc61UGRk1
📜Paper: https://t.co/xJkOlXmdbD
#AlphaFold #AlphaFoldMultimer #AlphaFold3 #ProteinComplexes #ProteinStructure #MSA #Interpretability #AntibodyEngineering #ComputationalBiology #StructuralBiology
Our review on AI for protein engineering is out now, about this too-fast-moving field full of hype and overclaim, yet one that is having a real impact on the world and can be described in a coherent manner without histrionics
https://t.co/woOWuyTV5R
BREAKING: An AI just wrote a research paper. Submitted it to a top science conference. Passed peer review. Nobody on the review panel knew it was AI.
The paper is called "The AI Scientist." Published last week in Nature. Built by Sakana AI in Tokyo, with researchers from Oxford and UBC.
Here is what it did — completely on its own.
It read existing scientific literature. Formed a hypothesis. Designed an experiment. Ran the experiment. Analyzed the results. Wrote the full academic paper. Then peer-reviewed its own work.
No human at any stage.
They submitted three fully AI-generated papers to a top ML conference under blind peer review. Human reviewers were told some might be AI, but not which ones.
One was accepted. It scored higher than 55% of human-authored papers at that same conference.
The accepted paper cost $15 in compute to produce.
Fifteen dollars.
Now here is the part nobody is talking about.
The team found a clear scaling law: stronger foundation models produce higher-quality research outputs. Better base model in, better science out.
Which means this gets dramatically better — automatically — every time a new model drops.
Right now it is limited to computational ML experiments. No biology. No chemistry. No physical labs.
For now.
What happens when the thing that discovers new science... is itself?
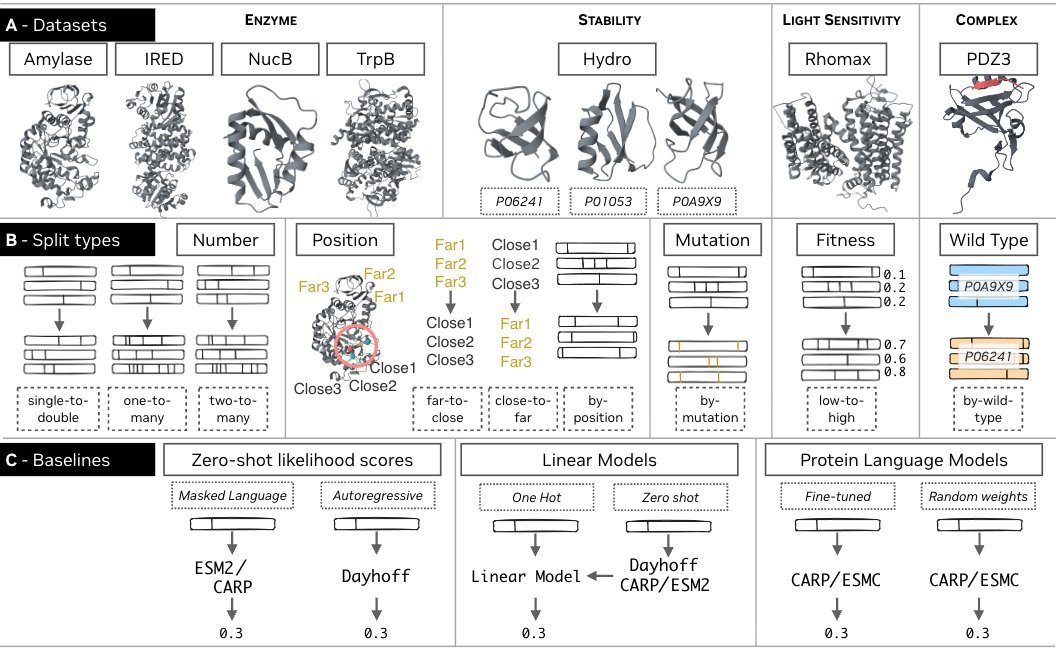
excited to release a new benchmark for protein fitness prediction: FLIP2
FLIP2 has 7 new datasets spanning enzymes, PPIs, and light-sensitive proteins, + splits designed to test generalization in realistic protein engineering settings
paper, data, code: https://t.co/6PQcn0YfGb
Mapping yeast's hidden metabolism with retrobiosynthesis and deep learning
Cells do far more chemistry than we know about. Beyond the reactions catalogued in textbooks and databases, enzymes routinely act on substrates they were never "meant" to process — a phenomenon called underground metabolism. In yeast, despite over two decades of genome-scale modelling, more than 90% of the metabolome remains unaccounted for. That gap isn't just a scientific curiosity: it means cell factories built to produce drugs, flavours, or biofuels are running on an incomplete map, with silent side reactions draining yield in ways we can't predict or control.
Ke Wu and coauthors address this systematically. Their pipeline extracts ~22,000 biochemical reaction rules from MetaNetX and MetaCyc, then applies retrobiosynthesis to expand all possible reactions across the full yeast metabolome — generating over 180 million candidate reactions. Two deep learning EC number predictors, CLEAN and DeepECtransformer, then annotate which yeast genes could catalyse each reaction, and the enzyme–substrate affinity model ESP ranks candidates and filters down to a tractable set. The result is Yeast-MetaTwin: 59,865 reactions, 16,244 metabolites, 1,976 genes — covering 92% of the yeast metabolome versus the 7% in the standard Yeast9 model.
The kinetic analysis is particularly illuminating. Comparing predicted Km and kcat values between known and underground reactions across multiple deep learning models reveals a consistent pattern: underground enzymes show roughly twofold higher Km values, but similar kcat distributions. Substrate affinity, not catalytic speed, is what separates promiscuous underground activity from core metabolism — a finding that puts quantitative grounding on a long-standing hypothesis.
The model also predicts by-products for heterologous products, and the authors experimentally validate two yeast genes — ADH6 and SFA1 — responsible for converting geraniol to geranial, a previously unidentified degradation pathway.
For engineering microbial cell factories, this kind of model changes what's possible in strain design. Predicting which endogenous enzymes will silently degrade your target compound — before you run a single fermentation — is exactly the kind of upstream intelligence that compresses development cycles in biotechnology and pharmaceutical manufacturing. As metabolome databases mature and deep learning enzyme annotation improves further, this approach generalises directly to human and bacterial systems, opening analogous opportunities in drug metabolism and microbiome engineering.
Paper: Wu et al., Nature Catalysis (2026) — Journal license | https://t.co/Vg2xv4cDQf
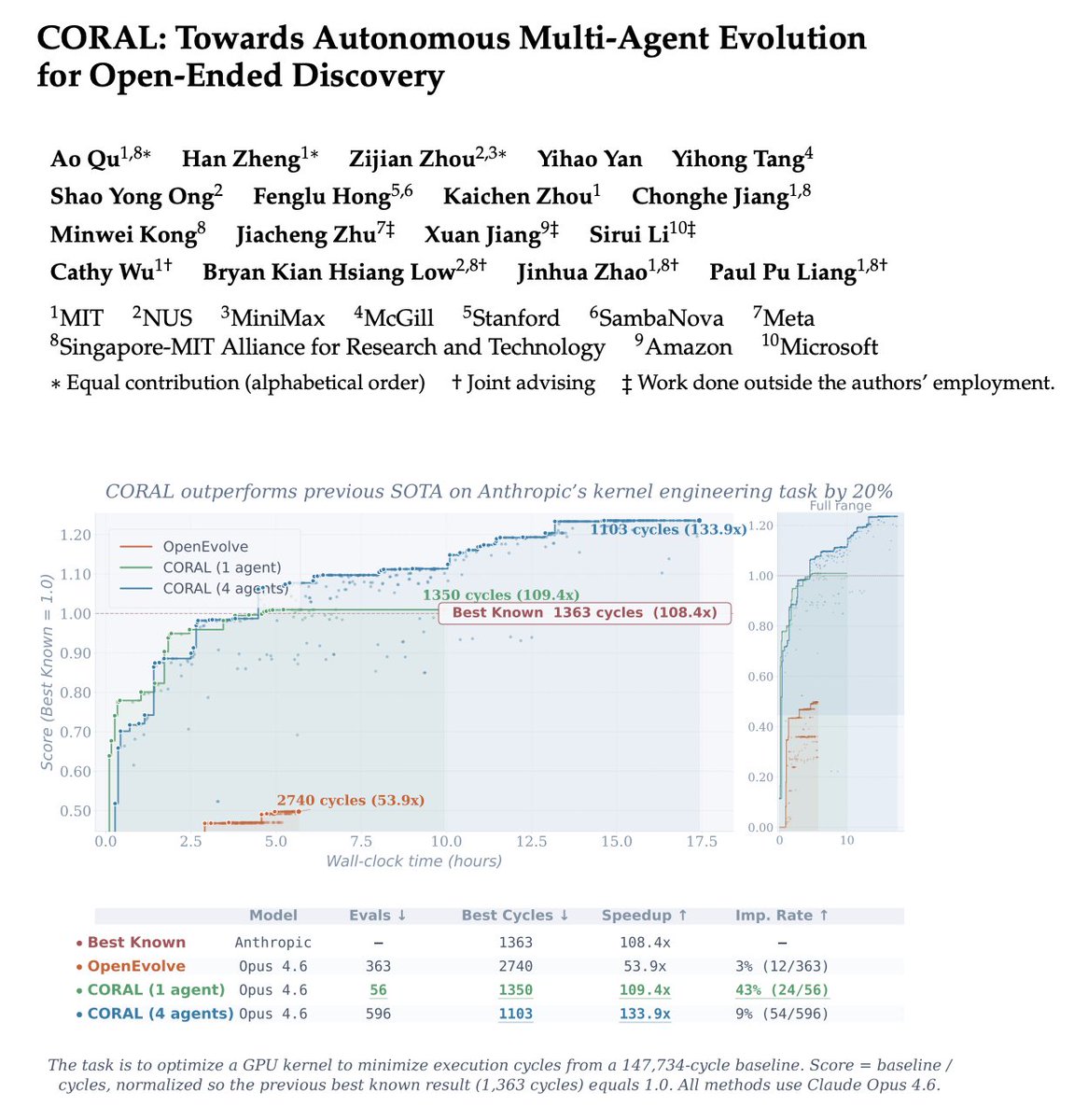
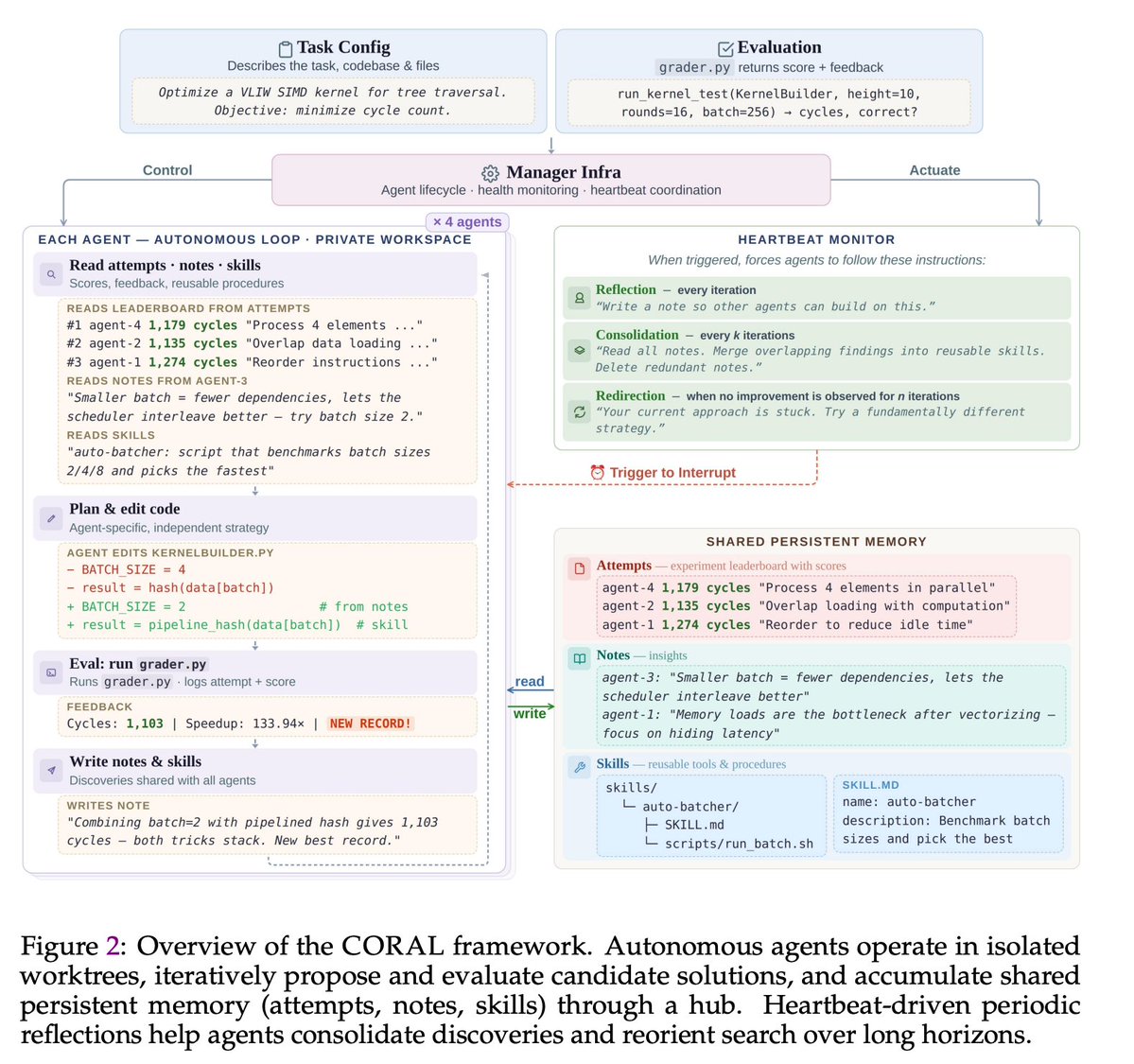
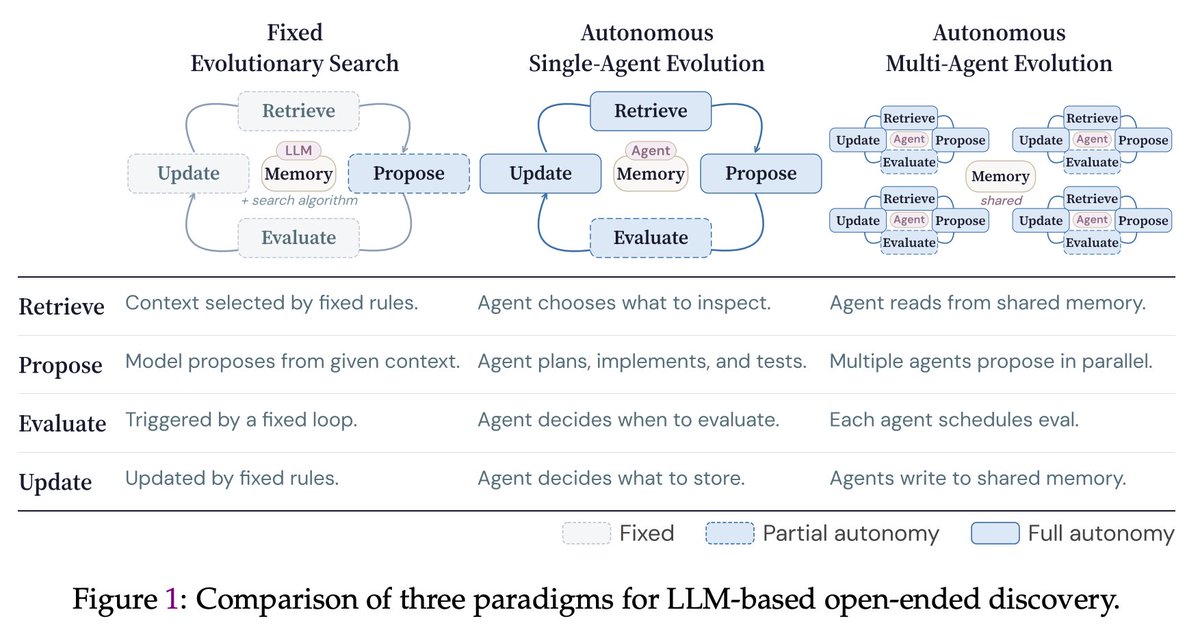
🚀The era of autonomous multi-agent discovery is arriving! @karpathy
🪸Excited to share CORAL, our new work on autonomous multi-agent systems for open-ended scientific discovery.
🙅♂️A key limitation of many current “self-evolving” frameworks is that agents still operate inside tightly constrained loops — they mutate solutions, but they do not truly decide how to explore.
In CORAL, we push toward genuine autonomy:
Agents decide
🔍 what to explore
🧠 what knowledge to store
♻️ which ideas to reuse
🧪 when to test hypotheses
🔥One of the most interesting findings:
A single autonomous agent already outperforms fixed evolutionary search, but the biggest gains emerge when multiple agents form a research community.
💪Over 50% of breakthroughs in multi-agent runs come from building on other agents’ discoveries. This suggests that knowledge reuse and collaboration are central to scalable automated discovery.
🏅Across 10+ difficult tasks in algorithmic discovery and system optimization, CORAL achieves state-of-the-art performance while improving efficiency by 3–10×.
📄 Paper: https://t.co/8ENJjgC5Xk
💻 Code: https://t.co/WjUJlG7B6p
💡AlphaXiv: https://t.co/TvheULeGgD
#agentic #llms #selfevolvingagent #multiagent #autoresearch #alphaevolve
Great work showing that protein language model embeddings can sometimes be indistinguishable from noise. This aligns very well with our observation in https://t.co/MJrsIoU6vB that motivated VESM / co-distillation.
Indeed, big knowledge gaps exist even within models of the **same family**, with almost identical architecture and trained on similar data: E.g., ESM2 and ESM1b each fail to detect well-conserved domains that the other recognizes as mutationally sensitive.
BREAKING: Anthropic Acquires 9-Person Biotech Startup For $400 Million
>be coefficient bio
>founded the startup 6 months ago
>build AI platform for biotech
>less than 10 employees
>acquired by anthropic for ~$400 million
> = $40+ million per head
Coefficient Bio was building an AI platform for biotech tasks: planning drug R&D, managing clinical regulatory strategy, identifying new drug opportunities
Team is joining Anthropic’s healthcare life sciences group led by Eric Kauderer-Abrams.
Anthropic is building specialized tools for industries that actually pay enterprise rates:
>software engineering
>cybersecurity
>life sciences
>healthcare
>finance
Meanwhile OpenAI is buying media companies to control narratives LMAO
What happens when you train a transformer on 123 million bacterial proteins
Bacteria have been fighting viruses for billions of years. To do it, they have evolved a remarkable diversity of antiphage defense systems — molecular immune machines that detect and destroy invading phages. Yet fewer than 250 such systems had been experimentally validated. A new study in Science suggests we've barely scratched the surface.
Mordret and coauthors asked a simple but powerful question: can language models trained on protein sequences and genomic context learn the "grammar" of bacterial immunity well enough to predict entirely unknown defense systems at scale?
They developed three complementary deep learning models. ALBERTDF adapts the ALBERT transformer architecture to treat genes as words and genome neighborhoods as sentences — learning defensiveness from genomic context alone, without any sequence information. ESMDF fine-tunes the ESM2 protein language model with LoRA adapters to classify proteins as defensive or non-defensive directly from amino acid sequence — trained on a dataset of 123 million proteins drawn from 32,000 bacterial genomes. GeneCLRDF combines both signals through contrastive learning: it teaches the model that a gene's identity can be inferred either from its sequence or from the genomic neighborhood where it lives. This joint embedding achieves 99% precision and 92% recall on held-out benchmarks — far outperforming each approach independently.
The models aren't just impressive on paper. The authors experimentally validated 12 antiphage systems with no prior link to immunity, in both E. coli and Streptomyces albus. Some carry canonical defense domains; others involve proteins with no known association to antiphage function whatsoever.
Applied to over 32,000 bacterial genomes, GeneCLRDF predicts 2.39 million antiphage proteins. Around 1.5% of a typical bacterial genome is devoted to defense — three times previous estimates — and more than 85% of predicted protein families have no prior link to immunity.
The implications are immediate. The predicted atlas — including ~23,000 candidate operon families — is a ready-made discovery pipeline for novel nucleases, molecular effectors, and antimicrobial mechanisms directly relevant to phage therapy and programmable biologics. Language models are turning the bacterial pangenome into an actionable resource.
Paper: Mordret et al., Science (2026) — Science license | https://t.co/CzpyZWPNbO
A lot of things happened in the AI x bio world these past few days. Here's what you might've missed.
☑️ An AI superforecasting system predicted a clinical trial would fail - before the results came in.
☑️ An AI caught what doctors missed across five brain diseases.
☑️ A foundation for interpretable virtual cell models - with wet-lab proof.
☑️ Three Ginkgo veterans say biology's bottleneck isn't better models - it's a missing design language.
☑️ 10,000 biomedical protocols in a simulation universe for AI lab agents.
Distilling the most confident mutation effect scores across different versions of the same protein language model results in a superior sequence-only variant effect predictor.
@tuanqdinh@vntranos
One protein. 10^150 possible DNA sequences.
We trained a transformer to pick the right one. 25 species. $165.
mRNA language models that learn context-dependent codon preferences from natural coding sequences.
Not frequency tables from the 1980s. Actual sequence understanding.
We're officially releasing the Adaptyv API, which gives you and your AI agents access to our wet-lab to test your proteins experimentally!
• Check out our demo and the docs here: https://t.co/3G81BmaNnb
• Check out how our partners @tamarindbio
and @phylo_bio have integrated the Adaptyv API into their platforms
We started Adaptyv with the idea that anyone should be able to test a designed protein, whether they have their own lab or not. Over the past three years we've tested tens of thousands of proteins from pharmas, AI for protein design companies, academic labs, alongside dozens of early-stage startups and individual researchers.
Until now, all of that went through our Foundry portal or also email threads and Slack channels. We think the process of testing a designed protein should be as simple as calling an endpoint, so we built an API around the same infrastructure those teams already use, to make everything as accessible as possible. As AI agents will do more and more scientific work, it's important to give them the tools to access real-world experimental validation.
To put it simply: AI can think but it cannot touch - we're giving AI access to the lab to validate experimental hypotheses.
Compressing the collective knowledge of ESM into a single protein language model
Predicting whether a genetic variant is harmful or benign is one of the most consequential tasks in computational biology. A mutation in BRCA1 or PCSK9 can mean the difference between a healthy carrier and a serious disease. Most top-performing variant effect prediction (VEP) methods get their edge by combining protein language models (PLMs) with 3D structure, multiple sequence alignments (MSAs), or population genetics — extra information that is expensive, incomplete, or potentially circular in clinical settings.
Tuan Dinh and coauthors ask a sharp question: are sequence-only PLMs fundamentally limited, or just under-exploited?
The key insight is that different ESM models — despite nearly identical architectures — have complementary blind spots. ESM2 reliably detects KRAB domains; ESM1b detects BRICHOS domains; neither catches what the other misses. Rather than averaging predictions (which dilutes rare signals), the authors select the minimum log-likelihood ratio across all models — the prediction most confident that a residue is mutationally sensitive. This signal then drives co-distillation of the entire ESM family into improved single models (VESM), through iterative rounds where models alternately teach and learn from each other.
The results are remarkable. VESM-3B, trained exclusively on unaligned sequences, matches or surpasses SaProt, PoET, TranceptEVE, and even AlphaMissense — a closed-source model trained on 3D structure, MSAs, and population allele frequencies. Critically, VESM maintains consistent performance across all allele frequencies, outperforming AlphaMissense precisely on the rare variants where clinical interpretation matters most. VESM scores also correlate quantitatively with continuous phenotypes in UK Biobank data, extending VEP from binary pathogenicity to quantitative trait prediction.
This is directly actionable: a sequence-only model at state-of-the-art accuracy removes hard dependencies on structural data or population databases, enabling scalable proteome-wide variant scoring — including for targets with no known structure, no deep alignment, and no prior clinical annotation.
Paper: Dinh et al., Nature Methods (2026) — CC BY 4.0 |
https://t.co/cboTWhQTi5