Discovering causes of rare disease through innovative technology and vigorous collaboration. @NIH-funded. Tweets by Data Coordinating Center @UWBiostat.
New in @Nature! “GREGoR: Accelerating Genomics for Rare Diseases” highlights how the GREGoR Consortium is advancing rare disease discovery through data sharing, multi-omics, and next-gen sequencing across 7,500 individuals in 3,000+ families.
🧬https://t.co/arZjAkYGz2
Read more about VizCNV, "an open-source platform that incorporates read depth and BAF to enable haplotype-aware CNV analysis" - a collaboration between the Baylor College of Medicine GREGoR Research Center and the Carvalho Lab Partner Group at PNRI.
Understanding the #genome isn't always about finding one wrong note. Sometimes it's about hearing the whole symphony. Learn how PNRI researchers are uncovering hidden causes of disease. @CMBCarvalho. Read more: https://t.co/95XMaR6u4M
#raredisease#geneticvariants
Read the full paper on bioRxiv to see how adoption of this unified data architecture accelerates rare disease research and discovery: 🚀 https://t.co/9WkuzJOhNs
#RareDisease#DataModel#GREGoR
Development and implementation of a common Consortium Data Model:
🧬 Enables large-scale rare disease research
🤝 Facilitate[s] cross-center data harmonization
💻 Enables data interoperability
I watched the recording of the recent @gnomad_project@GA4GH webinar on v4 to address common misconceptions about the gnomAD by @HeidiRehm@SamBaxterCGC and Katherine Chao. 👀 📺
Much of that was said is likely known to most who use gnomAD on a daily basis such as myself. Still, some interesting nuggets were in there. So, here are my unsorted notes, in case you find them helpful:
gnomAD is not a control group in the normal sense, but represents a reasonable reference set for severe diseases such as child-onset muscular dystrophy. It is best thought of as a general population dataset. It is NOT a healthy population dataset.
Included are individuals from case-control studies and biobank participants. Not included are cohorts specifically recruited for detection of early onset mendelian diseases to avoid any over-enrichment of causal variants.
Under US regulatory framework, there is no chance of any HPO terms that can be made available by a resource such as gnomAD. Because its freely accessible. No login required. Only sex, age and original study that the person was recruited in, is available to the gnomAD team. Only aggregated data can be shared. Not details on any specific participant.
The only description they have is, if a person is a "control" or "case" in the original study and that only refers to the small phenotypic window thats addressed in the original study (say schizophrenia or diabetes).
This is in contrast to @uk_biobank or @AllofUsResearch data. These datasets are not freely accessible - you only get to access them through their data portal where you need to login. But you get individual level data.
@HeidiRehm said it will be "a while" until gnomAD data is available on another genome build other than the current hg38. Conversations on e.g. T2T or pangenome graph type of future gnomAD versions have just begun.
Roughly half of the exome data made available in gnomAD v4 is from @uk_biobank. As it is a biobank that has a healthy volunteer bias, because volunteers tend to be slightly wealthier and healthier than the general population (see PMID: 28641372). Which actually means that the non-UK-biobank subset in gnomAD has a higher disease burden because it is more broadly recruited.
UK Biobank participants do have rare diseases, see PMID: 28641372
Individuals in gnomAD are also from case-control studies, such as schizophrenia, myocardial infarction etc. Thus the gnomAD team created subsets, such as non-neuro to filter out certain groups/study participants. See contributing projects here https://t.co/Rll5B28ZBt
At the current size of gnomAD in v4, any phenotype enrichment is less of a concern than in smaller previous versions. Simply because the dataset is 800k people.
Question - should a subset be used to apply e.g. the ACMG BA1 or BS1 criteria? - @HeidiRehm says v4 is best, when used as a whole dataset, because the more people, the better. If you want a max allele freq for a certain variant, then check ancestry subgroups.
All of gnomAD v4 can be considered as roughly "non-cancer" because all of the data from TCGA (The Cancer Genome Atlas Program) was removed due to QC. Which means, people in gnomAD can of course have cancer, but there is no enrichment of cancer phenotypes in v4.
Question - there seems to be higher freq of cancer variants in the non-UK-biobank subset. Why? - KC: Could be due to recruiting, since UK Biobank only recruited people aged 40-69 years old. They did find far lower rates of cancer in UKB than in the general UK population. Again, biobank participants are on average healthier.
@AllofUsResearch data will be inclued in v5 (+ another 800k individuals), which is "coming soon". Again, no phenotype data.
Question - what about CHIP variants? - @HeidiRehm: gnomAD has a full spectrum of age so likely no enrichment, but of course older people are in there with CHIP variants. There are flags on genes, to signal high rate of CHIP variants.
gnomAD does have age on many individuals included, but not all. This was information given by the original study. It is unclear to the gnomAD team actually what is meant by age in all of the different studies - e.g. time of inclusion into the study? Age when data was transferred to gnomAD? Other? The gnomAD team did not tinker with the age data, just used it as is, where possible and shares it in buckets in individual variant pages.
Question - are you planning to include the million variant program dataset? - @HeidiRehm - not yet included because of no easy access to it.
Since external sequencing data has to be reprocessed by the gnomAD team (quite expensive), only highly "valuable" datasets, meaning ancestrally diverse external datasets will be re-processed in future gnomAD. Simply due to cost-benefit.
gnomAD v4 is mostly saturated for common variants from european ancestries, so non-european ancestries are more "valuable". Here is the preprint for gnomAD v4 by @konradjk and team if you want to dive into that PMID: 41929314.
Question - why is #CFTR delta-F508 58x homozygous in gnomAD? @HeidiRehm - the team did a little research, as this is likely a bit too high for a general population. Turns out, a cohort from a clinic (important: not from UK Biobank) was included with a pulmonary focus.
https://t.co/v89wQnylOi
My former colleague @tomaeusTo also stumbled upon this #CFTR variant back in 2023 right after the release. See here for more details with a link to the gnomAD FAQ forum:
https://t.co/YJIZ1bOUVw
There are actually more than a few #CFTR over-represented variants in gnomAD, again likely due to the inclusion of that one pulmo clinic's cohort.
Question - what about LongReads? HR: @TalkowskiLab is working on a LongRead gnomAD dataset. It will happen. They are working on it. No date for release just yet.
There is a Canadian gnomAD dataset, not yet included in the primary gnomAD dataset. For now, it is available for download — with a browser interface coming soon. They actually want to provide phenotypes, but that is still "quite a few months out" (Jordan Lerner-Ellis).
https://t.co/ScPFH5Ug4b
https://t.co/M808TqEh3Q
https://t.co/SF8iN3NcKV
There is also a Singapore gnomAD dataset - probably not possible to access the data right now. So different groups can add their own wrinkles to these separate gnomad type datasets.
Recruiting more samples for gnomAD is ongoing. Federated gnomAD explained here: https://t.co/Xa9vLjU75e
@HeidiRehm points out there is also a gnomAD forum / discussion board. Please ask questions or feature requests. Link: https://t.co/bNUU98wOZ4
Thanks to the gnomAD on all their work on this invaluable resource. Couldn't do without it.
Hope this summary helps. 🙂
Abstract submissions are now open for AGBT Precision Health 2026 in San Diego, Sept. 14–16.
Submitting an abstract may result in:
• A plenary stage talk
• A poster flash talk
• A poster presentation in the outdoor poster tent
#AGBTPH26#PrecisionHealth
Genome sequencing (GS) is changing rare disease diagnosis. In a study of 10,000+ patients from Brazil, authors of @HGGAdvances latest article show that GS has strong potential to shorten the diagnostic journey—especially for underrepresented populations: https://t.co/fgTQxckQDc
📊 Spread the word about your tool or highlight your research results with other scientists doing genomics in the cloud! Submit an abstract today for a poster or talk at the AnVIL Community Conference 2026!
Deadline is July 15: https://t.co/gZNelrN8KP
Happy to share the latest papers describing a distinctive recessive neurodevelopmental syndrome resulting from the noncoding gene RNU4-2 with contributions from our BCM @GREGoR_research research center:
https://t.co/xBOmi9Doid
Is PCR bias clouding your gene editing analysis?
Join our webinar to see how amplification-free HiFi sequencing captures complex on-target and off-target edits that short reads miss. See how Uppsala University is leading the way.
Register: https://t.co/Ds1IL1oifi
#PacBio
Every discovery helps move the field forward.
Abstract submissions for #ASHG26 are now open. Add your voice. Submissions are due May 18. https://t.co/jbSLHa0sUV
National Public Health Week⚕️
During National Public Health Week, we recognize the impact of genomic medicine, driving more proactive, precise, and effective healthcare for all.
#NationalPublicHealthWeek#precisionhealth#precisionmedicine
Advancing #RareDisease research starts with #ResearchFunding strategies built for small populations and complex pathways. Hear how collaboration is driving progress at the #NORDSymposium. Grab your tickets at https://t.co/CeWGgwt15j.
What’s one of the biggest challenges in genomics today?
Lea Starita explains how variants of uncertain significance limit genetic medicine, and how IGVF is working to solve it using functional data and predictive models.
▶️ https://t.co/orIco85ksm
Incomplete penetrance in severe monogenic neurodevelopmental disorders is rare but probably under-appreciated. Happy to share our new preprint describing a surprising example of this phenomenon:
https://t.co/mDvqqyHNYM
Join us for a webinar on Thursday, April 2, at 10 am PDT.
Lea Starita, PhD, will demonstrate how the NHGRI's Impact of Genomic Variation on Function consortium developed methods to deliver clinically relevant information from experimental and predictive data.
Register here: https://t.co/Zm86Be9G9s
#VUS #genetictesting #CEUs #GeneChat
We are excited to announce the release of the Human Methylation Atlas Summary and Signals tracks for hg38 and hg19. The tracks display genome-wide DNA methylation profiles across 39 primary human cell types from 205 healthy tissue samples.
Learn more at https://t.co/GB3hxaYo1H
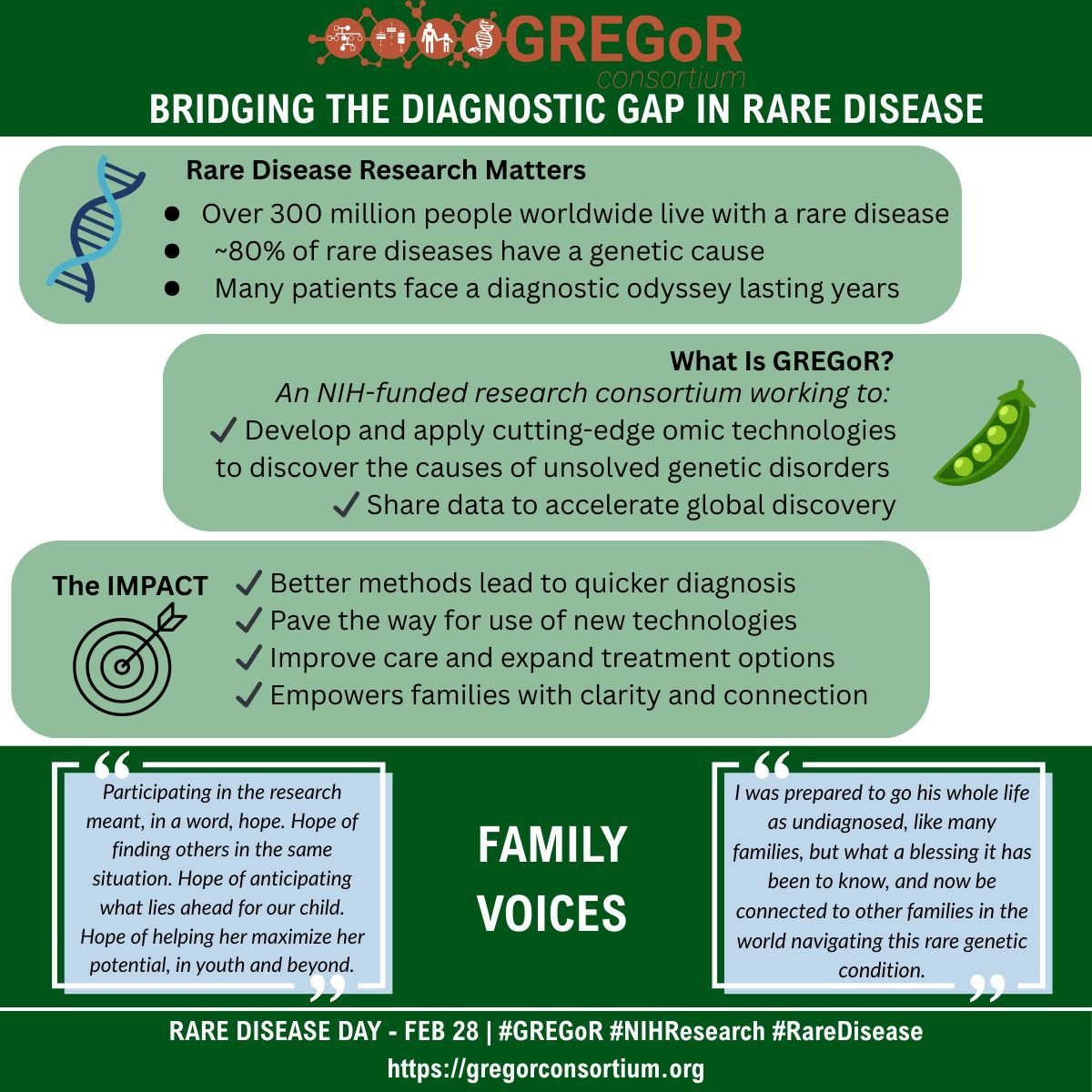
This Rare Disease Day 🧬, we share how GREGoR 🫛 bridges the diagnostic gap in rare disease by developing and applying cutting-edge technologies to discover the causes of unsolved genetic disorders and sharing data to accelerate discovery. 🎯
#GREGoR#NIHResearch#RareDisease
Excited to visit @viennabiocenter on March 10th to present our lates findings of Constellation/TruPath @illumina ! I did the PhD there so hopefully I can still find some familiar faces :) . I will talk about the application to rare diseases cases @BCM_HGSC@GREGoR_research