Introducing Open Molecules 25, a foundational quantum chemistry dataset including >100M DFT calculations across 83M unique molecules, built with 6B core hours of compute!
What does this mean for drug discovery, biology, and BioML?
1/
Excited to share our latest releases to the FAIR Chemistry’s family of open datasets and models: OMol25 and UMA! @AIatMeta@OpenCatalyst
OMol25: https://t.co/UhIDiFFzzN
UMA: https://t.co/eQ2wPIxbxY
Blog: https://t.co/VFaeynyZN7
Demo: https://t.co/Dj29ZfhBRO
The Open Molecules 2025 dataset is out! With >100M gold-standard ωB97M-V/def2-TZVPD calcs of biomolecules, electrolytes, metal complexes, and small molecules, OMol is by far the largest, most diverse, and highest quality molecular DFT dataset for training MLIPs ever made 1/N
A new flagship quantum chemistry dataset is out! Omol25 contains more than 100 million DFT calculations across an expanse of chemical space.
Collaborating with the FAIR team on this has been super fun!
https://t.co/kPJHqPdvwH
Announcing https://t.co/LWMnRVg9uP
Achira will usher in the next phase of AI for drug discovery building atomistic foundation models for biomolecular simulation to harness the explosive growth of available computation and the frontiers of physics-based synthetic data generation. Our models combine learning accurate AI-representations of physics with simulation, and embrace the paradigm of using inference-time computation to generalize beyond training.
Achira’s models will rival experimental accuracy with unprecedented experimental data efficiency, and help us turn drug discovery into engineering. Excited to be part of the journey with my long-time collaborator @jchodera , @zavaindar and this dream team.
Some news: I'm joining the founding team at Achira! We are building physics into AI to shape the future of drug discovery. If you're interested or just want to know more, hit me up!
https://t.co/JjbMjXlo1R
Presenting this work at NeurIPS tomorrow morning! I will be at NeurIPS from 12/11 to 12/14, let me know if you’d like to chat about AI for electronic structures, molecular dynamics, materials design, or the FAIR chemistry team!
Does equivariance matter at scale?
... When the twitter discourse gets so tiring that you actually go out and collect EVIDENCE :D
There has been a lot of discussion over the years about whether one should build symmetries into your architecture to get better data efficiency, or if it's better to just do data augmentation and learn the symmetries.
In my own experiments (and in other papers that have looked at this), equivariance always outperformed data augmentation by a large margin (in problems with exact symmetries), and data augmentation never managed to accurately learn the symmetries. That is perhaps not surprising, given that in typical setups the number of epochs is limited and so each data point is only augmented a few times.
Still, many "scale is all you need" folks believe that one should prefer data augmentation (or no bias at all) because eventually, with enough compute / data scale, the more general and scalable method will win (The Bitter Lesson). However, is data augmentation really more scalable?
Scalability: how fast the method improves with data and compute scale, and for how long it keeps improving. This is exactly what equivariant nets are good at!
We use transformers not N-grams for language, because they are more data efficient / scalable / better adapted to that problem domain.
Paraphrasing Ilya Sutskever: scale is not all you need; it matters what you scale.
In this latest work we decided to study the scaling behavior of equivariant networks empirically. As Johann explains in the thread below, we confirmed that equivariant networks are more data efficient. Interestingly, we were also able to confirm the intuition that in principle, the network should be able to learn the symmetry as well! When data augmentation is applied at sufficient scale, you get the same sample efficiency benefits as equivariance. HOWEVER: you need to do a huge number of epochs (which people don't do in practice), making equivariant networks more efficient / scalable in terms of training compute.
So equivariant networks allow you to get the statistical benefits without paying the computational cost.
The takeaway for me is that if you are working on a problem with exact symmetries, and are working on it because it is intrinsically important (climate, materials science / chemistry, molecular biology, etc.) rather than as a stepping stone to a more general problem (where the inductive bias could fail), then equivariant nets are still a good candidate in the age of scaling laws.
Awesome work @johannbrehmer@pimdehaan Sönke Behrends!
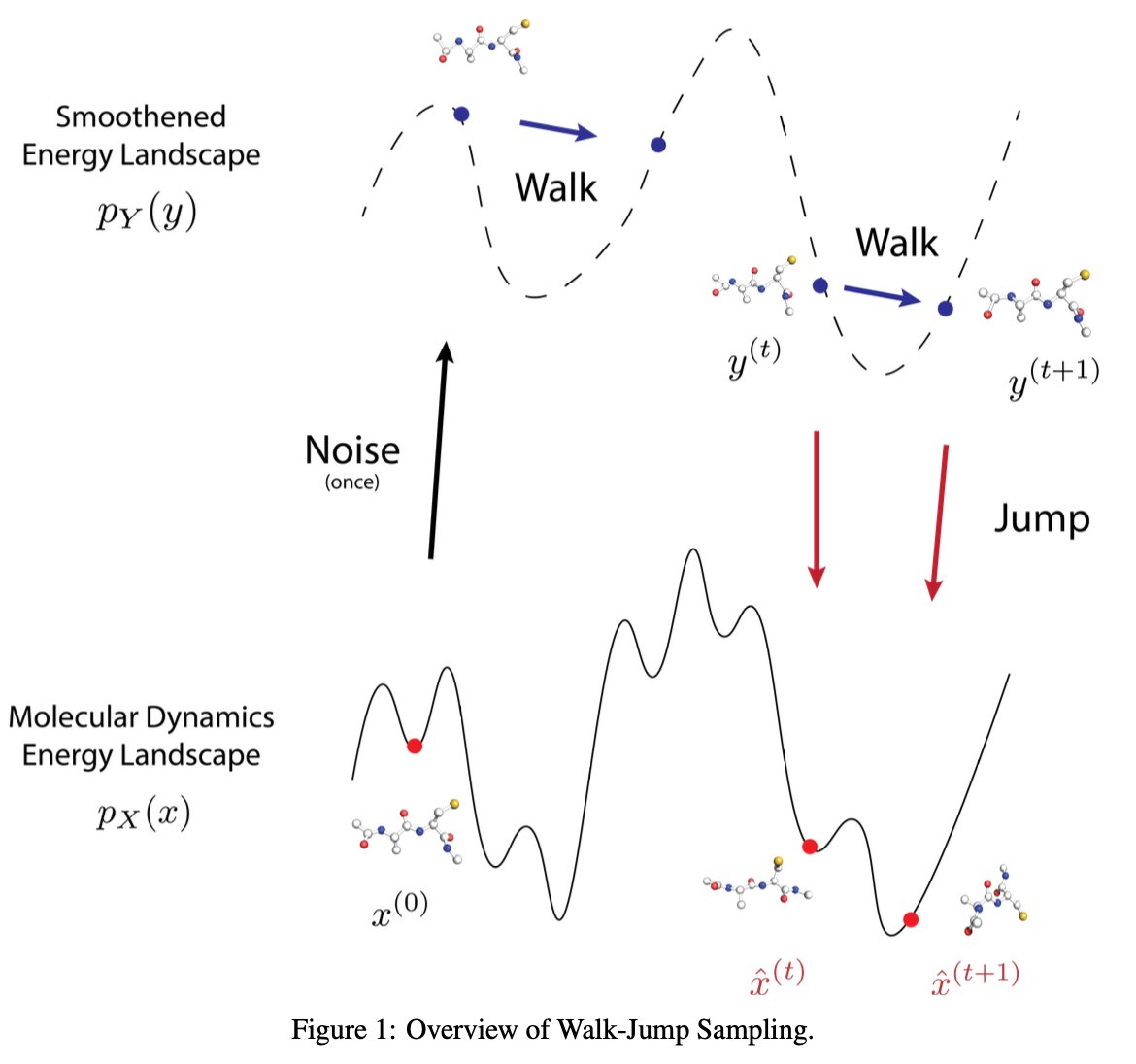
JAMUN: Transferable Molecular Conformational Ensemble Generation with Walk-Jump Sampling @PrescientDesign
• JAMUN introduces a generative model based on Walk-Jump Sampling (WJS) to efficiently generate molecular conformational ensembles, outperforming traditional molecular dynamics (MD) in speed.
• The key innovation lies in using WJS with SE(3)-equivariant denoising networks, preserving the physical priors from MD data while improving transferability to unseen peptides.
• JAMUN’s approach addresses the limitations of short time-steps in MD by sampling from a smoothed manifold, offering faster convergence to stable conformations.
• Unlike other ML methods, JAMUN demonstrates transferability, generating stable conformations for peptides outside its training set with remarkable fidelity.
• Ablation studies highlight that JAMUN generates conformations at a fraction of the time required for MD, providing orders of magnitude speed improvements in ensemble generation.
• With applications in drug discovery, JAMUN offers new opportunities to explore cryptic pockets and antibody design through rapid generation of conformational ensembles.
@JoshRackers@bodhivani@BigAmeya
📜Paper: https://t.co/NzIIq7AZ5v