When I start a computational biology project these days, I set up a git repo and a LaminDB instance. It lets me do a lot more, in a reasonable time, in a reproducible way. That's a rare combination in this field. I recently made a short blogpost about it: https://t.co/AdUWmH67TN
When spatial datasets accumulate across experiments and technologies, managing, querying, and training models on them becomes a major challenge. To address this, we built support for scverse's SpatialData format into LaminDB, enabling cross-dataset queries, dataset validation, and lineage tracking.
The main challenge was extending pandera-based schema validation to the complicated structure of SpatialData; Parquet and AnnData are easier!
Blog: https://t.co/pmpT2A6uOy
Code: https://t.co/tb6t2FJ7tt
With @LukasHeumos and many others!
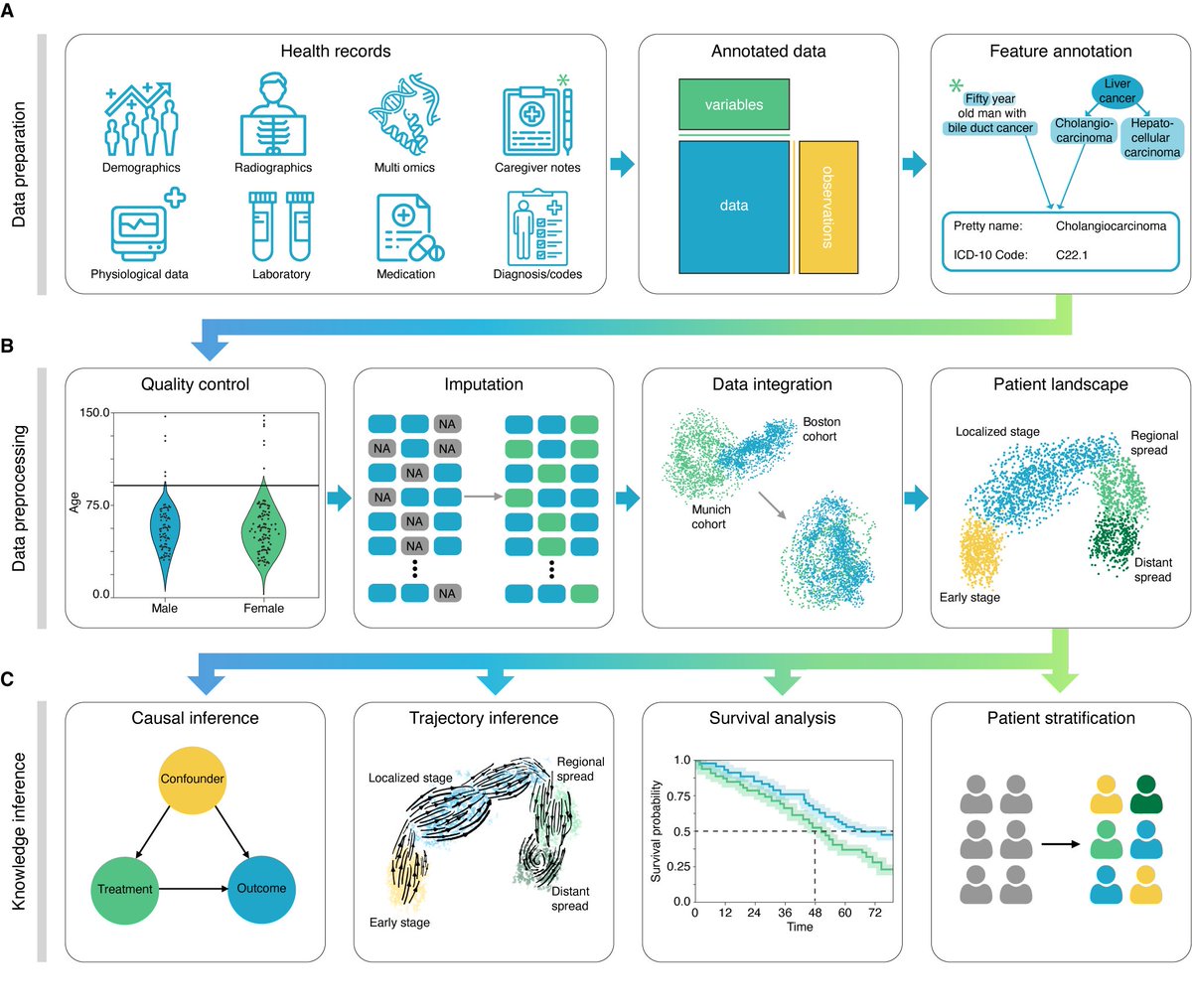
Happy that our ehrapy toolbox for the exploratory analysis of electronic health records is out today @NatureMedicine! It enables early QC&imputation at scale, visualization and downstream clustering & patient trajectory learning.
https://t.co/PTqJ5P4C8X https://t.co/CJRIsM8jzi
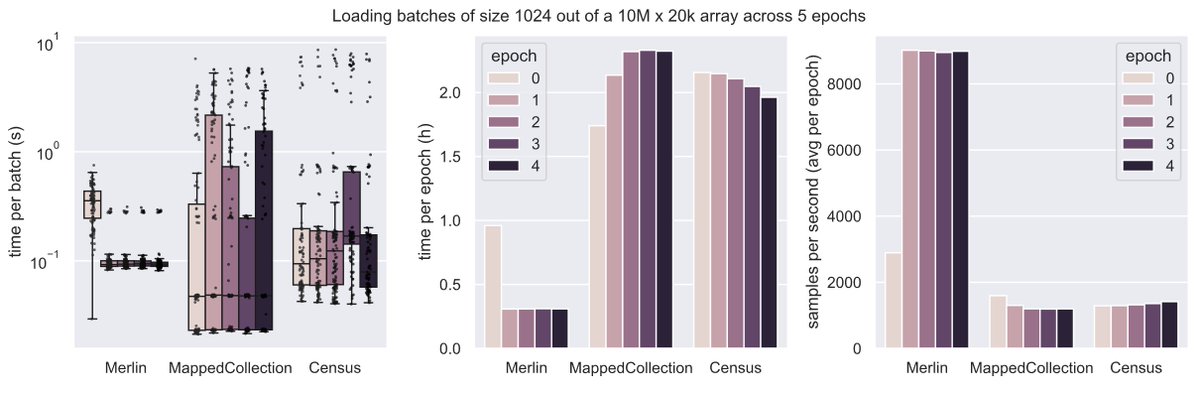
@_canergen@falexwolf >Are some dataloaders CPU limited or all disk speed limited?
I don't think we hit disk speed limits with any dataloader, MappedCollection is just slower because during the random sampling (training mode) it pulls individual indices instead of whole chunks like Merlin does.
@_canergen@falexwolf Hi, @_canergen ,
>Have you tried load_sparse_tensor similar to scVI?
MappedCollection samples indices one by one randomly and then PyTroch DataLoader collates them into a tensor, it really loads neither sparse nor dense whole tensors. And Merlin stores and samples dense arrays.
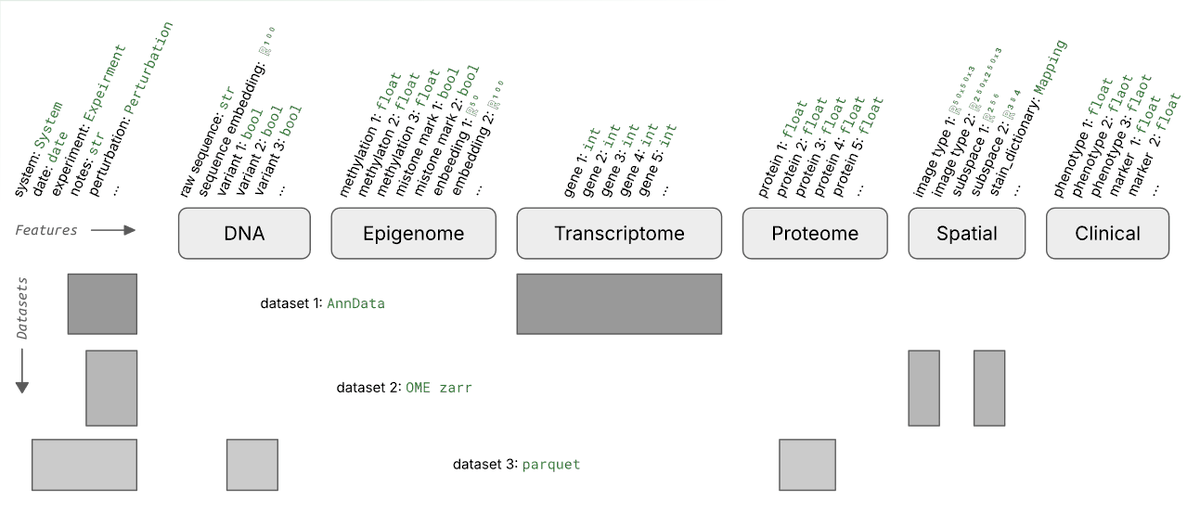
What's a good way of organizing scRNA-seq data for training foundation models?
Say you run 1k experiments and each measures counts for 1M cells with varying metadata and orthogonal data.
Storing these data in one gigantic array isn’t exactly easy.
We wondered whether it’s necessary to train foundation models and found 3 setups that made sense to us.
https://t.co/4p6g3iRbpE
I'm super excited to announce our new framework for exploratory electronic health record analysis "ehrapy". Although analysis is standardized for single-cell by seurat, bioconductor and scanpy, EHR analysis was until now the wild west. https://t.co/eOfJ0UaPum
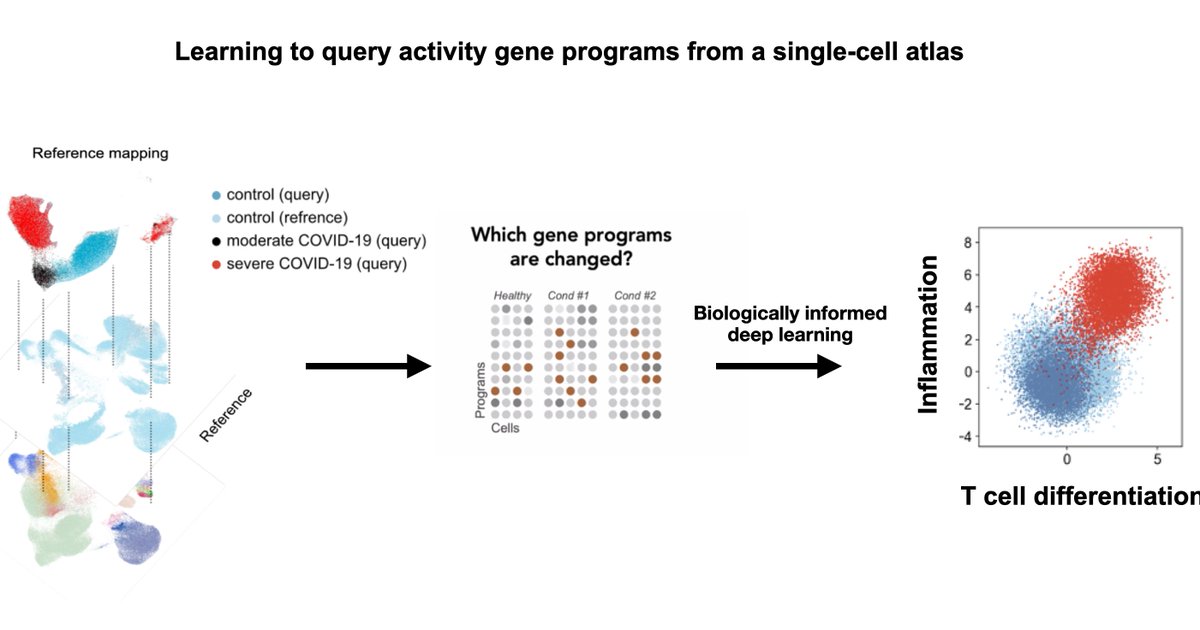
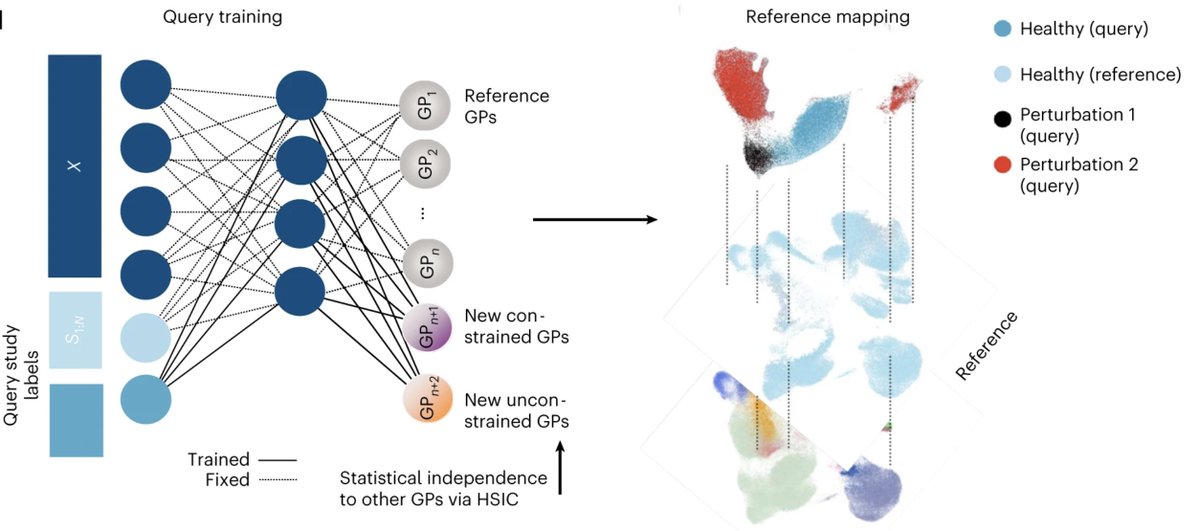
(1/4) Thrilled that expiMap is now published in
@NatureCellBio. It learns the activity of gene programs for a query single-cell data in a reference atlas while enabling learning novel gene programs (e.g., new cell states, disease)! https://t.co/iloivSId95
Excited to finally see ExpiMap out @NatureCellBio - led by @Mohlotf & Sergei Rybakov, we inform single-cell embeddings by pathway priors (+ newly-learnt ones). This allows for biologically understandable components in the latent space and program queries. https://t.co/czvxbbr4Eu
We are very excited to announce scverse (https://t.co/B4lDGfWDPQ), a new consortium around the core Python packages for single-cell omics data analysis. scverse is a cross-lab effort to ensure the longevity and interoperability of the single-cell analysis ecosystem in Python.
AnnData 0.8.0 is out!
New features include:
* Refactored IO, including low level access and support for new datatypes
* Out of core pytorch data loaders
* AnnDatas without an X value
Check out the full release notes here: https://t.co/yDUqf9LVbL
(1/11) Excited to share our new approach to learn gene programs (GP) activity from single-cells "biologically informed deep learning".We add prior knowledge while learning new cellular circuits, going beyond data integration and towards interpretability.https://t.co/uaIYvwcUnj
@deleeuw_jan sorry bo bother you with it, but is it still possible to get a pdf version of your book "Block Relaxation Methods in Statistics"? The link https://t.co/tGbm5UvHro doesn't work for me, it says "You don't have permission to access /bras/_book/_main.pdf on this server".