Today we're announcing ESMFold2, an open scientific engine to power prediction, design, and discovery across protein biology.
The new model delivers state of the art performance on protein interactions, especially antibodies, a critical modality for therapeutics.
We have designed and validated miniprotein binders and single chain antibodies across five therapeutic targets that are important in cancer and immunology. We are seeing very high success rates, and affinities at levels consistent with therapeutic activity.
We’re also releasing an atlas of 6.8 billion proteins, and 1.1 billion predicted structures.
ESMFold2 is built on a state of the art language model that has been trained on billions of protein sequences.
A world model of protein biology emerges through language modeling.
We’ve used the techniques of mechanistic interpretability developed to understand large language models to understand the concepts ESM uses to represent proteins.
The model’s representation space has a compositional organization of features across scales, levels of complexity, and abstraction, that reflects and mirrors the understanding of protein biology developed through a century of empirical science.
This understanding emerges without prior knowledge, just from language modeling of protein sequences.
Language models are becoming a powerful substrate to understand and program biology.
The design of protein interactions is one of the most fundamental problems in biophysics, and has critical implications for the discovery of new medicines. A simple gradient based search with the model was able to discover high-affinity protein binders.
I'm excited by the potential this has to accelerate basic science and the understanding of proteins. And especially for the new avenues it opens up for therapeutic design and medicine.
Still incredible that the DeepMind documentary has footage of exact moment Demis is told that AlphaFold can “easily” predict all known (1-2B) protein sequences “in a month” and he says to do it.
Then, it shows the moment AlphaFold is released to the world.
@vntranos I am missing ESM-IF in your approach, technically is a PLM also. You also have models that accept structural input. Is there any particular reason why was it not consider?
Huge advance towards the idea of using multiple PLMs as a roundtable of experts to clasify protein variants reliably.
"Sequence is all your need" could be also applied directly to other fields like enzyme engineering 😉
Excited to share that our latest work building on ESM is now published in @NatureMethods:
A single, sequence-only protein language model achieves state-of-the-art variant effect prediction, surpassing hybrid approaches that use MSA, 3D structure, or population genetics data.
https://t.co/MJrsIoU6vB
Fast sampling of protein conformational dynamics @ScienceAdvances
1. Sauer et al. show that the key collective variables (CVs) needed to drive enhanced sampling of protein conformational transitions are encoded in anharmonic low-frequency vibrations, and these CVs can be extracted from short unbiased MD without any prior knowledge of the transition.
2. Core idea: use FRESEAN (frequency-selective anharmonic mode analysis) at (near) zero frequency to isolate collective motions with minimal restoring forces—i.e., “paths of least resistance” for conformational change—avoiding the limitations of harmonic/quasiharmonic normal modes in the low-frequency, diffusive regime.
3. Practical pipeline: run 20 ns unbiased all-atom MD, align trajectories, coarse-grain to a 2-bead-per-residue representation (1 for Gly), compute velocity time-correlation matrices, Fourier transform to frequency domain, then take eigenvectors at zero frequency. Modes 1–6 correspond to translation/rotation and are discarded; modes 7+ capture internal anharmonic low-frequency vibrations.
4. Reproducibility is a central result: across 5 independent 20 ns replicas per protein, the low-frequency modes (especially the 2D subspace spanned by modes 7–8) are consistently recovered, unlike PCA/quasiharmonic modes whose replica-to-replica agreement remains poor even with much longer trajectories.
5. Enhanced sampling step: use modes 7 and 8 as CVs in well-tempered metadynamics (100 ns per run; reported as <24 hours on a single GPU). Across 5 proteins × 5 replicas, 22/25 runs (88%) sample known “closed↔open” transitions within 100 ns; extending to 160 ns yields full sampling for all replicas.
6. Benchmark set spans diverse challenges: HEWL (disulfide-stabilized), HIV-1 protease (homodimer), MCL-1 (allosteric/druggable dynamics), ribose-binding protein (multi-domain hinge motion), and GDP-bound KRAS (switch-region dynamics). The same FRESEAN-to-metadynamics protocol is applied across all systems.
7. Free-energy landscapes (FES) become both fast and statistically controlled by running 20 parallel metadynamics replicas (20 × 100 ns) using the same FRESEAN CVs: single-run uncertainties are typically < ±10 kJ/mol, and averaging reduces standard error to < ±3 kJ/mol, enabling reproducible thermodynamic ensembles rather than just qualitative transitions.
8. Comparison to “hand-crafted” geometric CVs from prior literature is informative: biasing along FRESEAN modes often follows lower-free-energy transition routes and tends to keep sampling within the native folded ensemble, whereas geometric CVs can push systems into partially unfolded high-entropy states (most notably KRAS when biased by residue–residue distances).
9. The authors quantify cross-CV reweighting fidelity using Shannon entropy and Bhattacharyya coefficients: on average, ensembles generated by biasing along low-frequency vibrational CVs preserve at least as much (often more) information when reweighted into geometric-variable space than the reverse, supporting the claim that these vibrations are broadly suitable, system-agnostic CVs.
10. Implication for computational biology/ML: the method enables high-throughput generation of conformational ensembles and FESs (including mutants/conditions), helping address the dataset bottleneck for next-generation sequence→structure→dynamics models beyond single static folds or single thermodynamic states.
💻Code: https://t.co/Ve9sGLVFXE
📜Paper: https://t.co/tcpFIhuUhs
#MolecularDynamics #EnhancedSampling #Metadynamics #ProteinDynamics #FreeEnergy #ComputationalBiophysics #CollectiveVariables #FRESEAN #GROMACS #PLUMED
Cool analysis, being able to test the limits and bias of our tools is an important first step into being able to reliable discriminate true negatives from our designs.
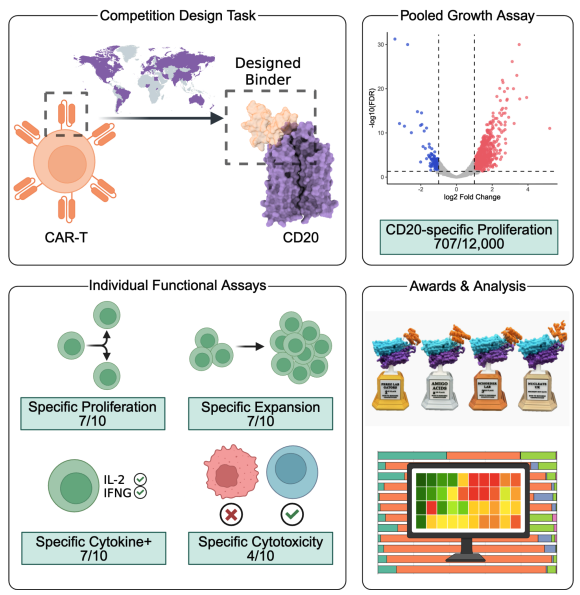
The results are finally in! 🏆💻🧬
I'm thrilled to announce that the manuscript for the Bits to Binders protein design competition is out on bioRxiv! Here's a summary of our findings, including some simple criteria that nearly *double* success rates when applied as a filter 🧵
I don't like this idea of keeping the research secret to avoid people looking at it. I understand you don't have any pressure to show the progess, but my career depends solely on my work and my ideas. Why are PIs like that? #AcademicTwitter
Mechanisms of AI Protein Folding in ESMFold
1 Researchers have mapped out exactly how ESMFold computes protein structures, revealing a clear two-stage computational pipeline inside the folding trunk that transforms amino acid sequences into 3D shapes.
2 Using activation patching—a technique borrowed from language model interpretability—the team showed they could transplant structural motifs between proteins by manipulating internal representations, successfully converting alpha helices into beta hairpins and vice versa.
3 The first stage (blocks 0–7) propagates biochemical signals from sequence to pairwise representations, where features like electrostatic charge get encoded in linear, steerable directions. The second stage (blocks 25–35) develops spatial geometry, with pairwise representations functioning as distance maps that directly control the final structure.
4 A striking finding: charge information is linearly encoded and causally influences folding. By steering sequence representations toward opposite charges on facing strands, the researchers could induce hairpin formation through electrostatic complementarity—demonstrating that molecular physics is not just learned but actively used during inference.
5 The pairwise representation z acts as a geometric blueprint: linear probes predict distances with R² ≈ 0.9 in late blocks, and scaling z proportionally scales the output protein structure, confirming its role as a distance map that the structure module reads to generate coordinates.
6 This work establishes the first mechanistic understanding of protein folding trunks, showing that structural decisions can be localized, traced, and manipulated with strong causal effects—opening paths for interpretable protein design and targeted intervention in folding models.
💻Code: https://t.co/SSCfRO286m
📜Paper: https://t.co/1VvG7dskg6
#ProteinFolding #MechanisticInterpretability #ESMFold #AlphaFold #StructuralBiology #AIforScience #Bioinformatics #DeepLearning #ProteinDesign
Why academia is full of bad people?? I really don't understand how so many assholes reach a professor position. They are just damaging the system and making people with truly passion not wanting pursue it #phdhelp
Be the scientist who provides thoughtful and critical feedback to strengthen an idea without changing the idea. We need creative minds, not clones of thought. Mentors who foster creativity can have an enormous impact on the scientific enterprise: self-awareness is key ♥️ 🔑
Doing science today feels like playing a video game on legendary difficulty.
With shrinking budgets, extreme publication fees, subjective rejections, and daily lab chaos, the path of a scientist is tougher than ever.
At least we have Parafilm to hold it all together.
Stay strong!
@AdrianoAguzzi Yes intellectual property protection. All the departments are saying you can't upload data of on-going research to any of this websites. So I understand you can run the code locally, but sharing it with openai may cause problems with the university