Refining catalyst–adsorbate interatomic potentials with transfer learning in ænet-PyTorch
From optimizing catalyst interfaces to extending molecular dynamics (MD) simulations, linking broad chemical knowledge to specific adsorbate systems often poses challenges in materials research. While large-scale data repositories can help, constructing accurate machine learning potentials (MLPs) for adsorbate-catalyst complexes still requires significant computational resources, especially if only a small custom data set is available.
A recent paper by An Niza El Aisnada and coauthors proposes a transfer learning strategy to build stable MLPs under tight data constraints, particularly for catalyst–adsorbate systems. Leveraging the Open Catalyst 2020 (OC20) database—a substantial collection of diverse catalyst configurations—they pretrain MLPs on carefully selected OC20 subsets. By transferring the pretrained models to a smaller target data set (only a few hundred ab initio references), they achieve robust energy and force predictions. Notably, these transfer-learned MLPs remain stable for hundreds of picoseconds of MD simulation on Cu–Au/water cluster systems, whereas models trained only on limited local data fail much sooner.
They explore two main approaches for selecting relevant subsets from OC20: (1) random sampling to mirror the original database broadly, and (2) filtering by chemical environment (for example, focusing on Cu–Au). The pretrained MLPs, once transferred, exhibit significant improvements in force prediction and MD stability—even though raw RMSE metrics in smaller data sets do not always reflect such gains.
A key component of their workflow is the “ænet-PyTorch” framework. Originally, the Atomic Energy Network (ænet) was a C/Fortran toolkit for ANN-based MLP construction. In this updated PyTorch extension, parallelization and GPU acceleration are harnessed for efficient training, allowing the incorporation of both reference energies and forces. Through transfer learning, a user can import a pretrained model (from large data sets), then fine-tune it on domain-specific references to achieve both accuracy and scalability.
Beyond a simple methods comparison, the authors emphasize pragmatic insights—such as the importance of CV-limited data curation, the synergy of domain-focused subset selection (e.g., focusing on Cu–Au to boost transfer success), and the pitfalls of relying on single scalar metrics like RMSE. They illustrate how data set sizes and neural network hyperparameters (for balancing energy vs. forces) drive generalizability in practice.
Paper: https://t.co/2xajmrOr3u
One of the largest materials datasets and SOTA ML potentials are open-sourced by FAIR Chemistry.
Immensely proud to be part of the team that delivers open science and catalyzes community involvement and advances in this field.
https://t.co/IhWYAJZFzk
BREAKING NEWS
The Royal Swedish Academy of Sciences has decided to award the 2024 #NobelPrize in Physics to John J. Hopfield and Geoffrey E. Hinton “for foundational discoveries and inventions that enable machine learning with artificial neural networks.”
Very interesting roundtable on "How to best combine AI and physical modelling to accelerate discoveries with societal impact?" moderated by @nicola_marzari, with Laura Toni, @adrian_roitberg & @MicheleCeriotti at the #CECAM55 conference!
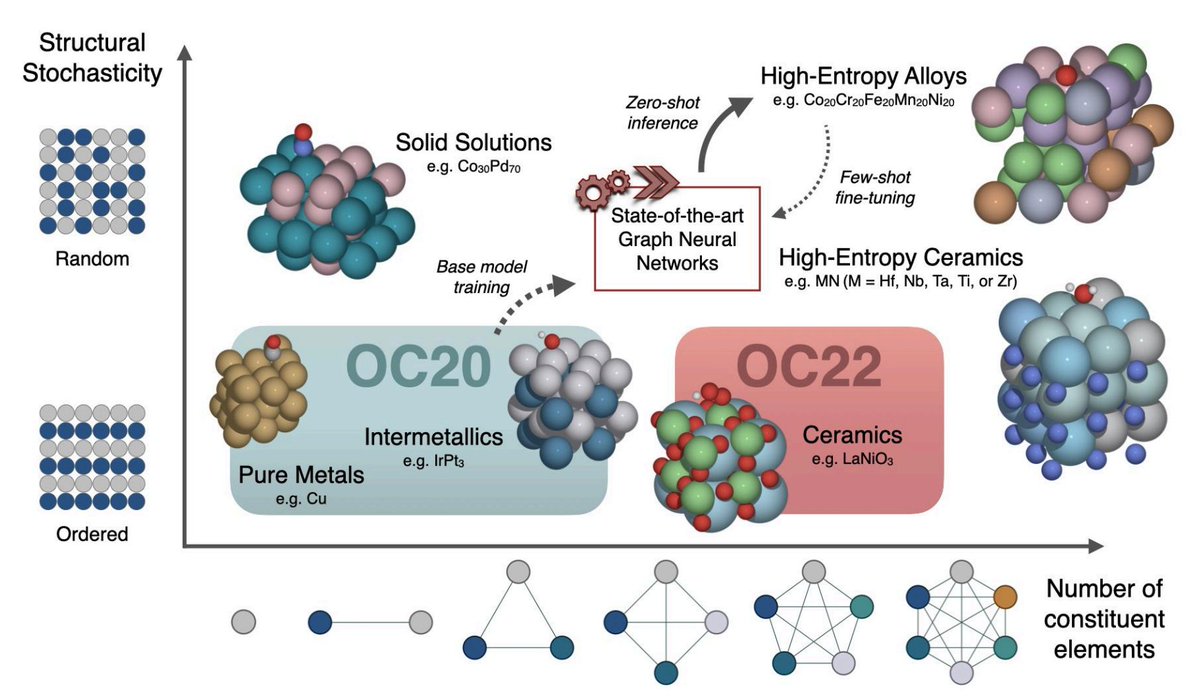
Paper shared on arXiv showing catalyst AI/ML models trained on datasets like @OpenCatalyst can generalize to solid solutions like high entropy alloys (HEA)! This is exciting because the design space of HEAs (with >5 components) is combinatorially large.
https://t.co/DHcdrZIRPR
Glad to share our recent work on modeling the cation-disorder in halide superionic conductors. 🔋
An interesting study integrating cluster expansion Monte Carlo & machine learning interatomic potential molecular dynamics. 👇
Bye, Viennese Schnitzel; hello, California rolls! I've just moved to @UCB_Chemistry. I'm looking forward to new challenges and collaborations. And I thank the tremendous support and lovely colleagues from @ISTAustria over the past few years.
SUNER-C Consortium Meeting at @UniUtrecht (1)
Last week we've organized the SUNER-C 4th Consortium Meeting. On Thursday the event took place in the Speelklok Museum in the city centre; on Friday we were in the David de Wiedbuilding, where there was a.o. a lab tour.
@sunergy_eu
We proudly present our 524 page book on equivariant convolutional networks.
Coauthored by Patrick Forré, @erikverlinde and @wellingmax.
https://t.co/y9YBpqhyLG
[1/N]
This looks amazing: capturing carbon from seawater and generating hydrogen as a byproduct. Given the rate the climate is changing I am sometimes wondering why we are not pouring billions into these projects and put ITER / LHC on the backburner for a while. https://t.co/kQ0TOlAlCi
David Reichman, JCP Associate Editor and Professor of Chemistry at Columbia University, wishes JCP a Happy 90th Birthday 🎈 - a journal he has looked to and respected since he was an undergraduate. @ChemColumbia
⚡️🔋☀️Attending the 2nd Edition of the NWO NERA (NL) Symposium, '#EnergyTransition - Collaborating towards a Sustainable Future' I think our ML/AI models could also be useful 😊#NWONERA Come see our poster & look forward to opportunities to work together ☀️@NArtrith@Peter2Ngene














































![maurice_weiler's tweet photo. We proudly present our 524 page book on equivariant convolutional networks.
Coauthored by Patrick Forré, @erikverlinde and @wellingmax.
https://t.co/y9YBpqhyLG
[1/N] https://t.co/Xf7DzkZx4I](https://pbs.twimg.com/media/F8tveafWoAANtjt.jpg)






















