Assistant professor at Dartmouth College. PhD@Yale, PostDoc@UChicago. Disease genetics, genomics, statistical genetics, data science. We are hiring! She/her.
Tenured or tenure-track biostatistics faculty position at the rank of Associate or Full Professor available @BioMedDataSci and Dartmouth Cancer Center (@DartmouthCancer) at Dartmouth’s Geisel School of Medicine (@GeiselMed)! Learn more and apply here 👉 https://t.co/2WjaK8Q7Z9
Our software is available at https://t.co/qW0jgUNQqB. Many thanks to my postdoc advisors @xinhe@mstephens999 co-first author @wescrouse, Sheng Qian and Kevin Luo! Please try our R package and let us know your feedback. Thanks! n/n.
We then applied our method to a few commonly studied traits (IBD, SBP, schizophrenia) and revealed novel candidate genes. Below is an example of a novel candidate gene (UBE2W) cTWAS identified for IBD, whose association falls below standard TWAS cutoff. 5/n
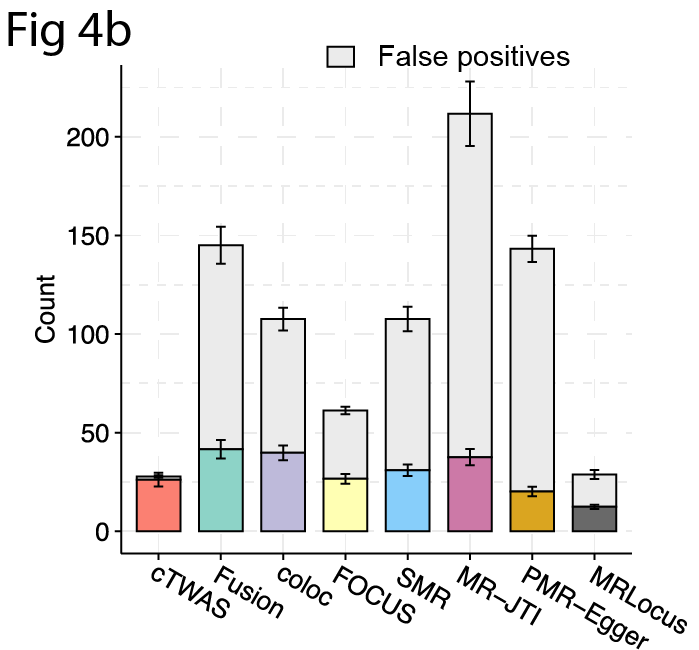
When applying cTWAS on real data, it identified much fewer genes than TWAS, the majority of the TWAS false positive findings were due to confounding by nearby causal variants. 6/n
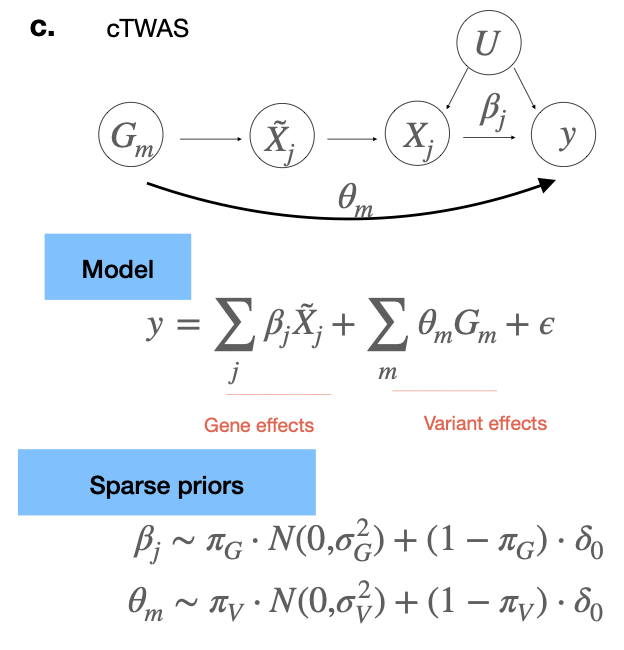
cTWAS solves this problem by including **all** nearby genes and variants in a Bayesian fine-mapping model, which effectively let gene and variant effects compete to explain the association signal. cTWAS has a well calibrated FPR, without much loss in power. 4/n
Glad to see our paper, with @xinhe2@mstephens999@wescrouse Sheng and Kevin, is out today at @NatureGenet! https://t.co/hMfTQYsGuK. We developed a new statistical method, causal-TWAS or cTWAS, to reliably identify causal genes in genome-wide association studies (GWAS). 1/n
nearby causal variants or eQTLs of causal genes of the trait can be correlated with the gene under test, thus serving as confounders in the test and resulting in false positives (FP). In simulations, we show previous methods can be severely inflated with FP ( FPR > 50%). 3/n
Many methods have been developed to leverage eQTLs to nominate candidate genes of complex traits, including TWAS, colocalization, mendelian randomization (MR) based methods, etc. However, they all suffer from a key problem: 2/n
@mstephens999@Sun_Y_Lee It should work, though the current version only takes two groups of variables as covariates, variant genotypes and imputed expression. Wes sheng and me are working to release a new version which can take multiple groups of variables, to include e.g protein expression.
I missed tweeting this when posted in Dec. Our attempt to ameliorate some problems with confounding in TWAS. Key idea is to compete (imputed) gene expression and SNPs against one another as explanations for an asssocation. https://t.co/zTnGPqLPAS
Molecular & Systems Biology at the Geisel School of Medicine at Dartmouth is excited to recruit Assistant/Associate Professors to our department. Find information on the breadth of our search and our commitment to faculty diversity at https://t.co/9vvKi0jzKf @GeiselMed@Dartmouth
The University of Chicago, Department of Human Genetics, is searching for a new computational faculty.
https://t.co/nBhQKsty0i
We are a small, friendly department doing top-quality science. Come join us! (Please RT)
Rockefeller's president Richard Lifton, who pioneered the use of genomics to identify the basis for diseases, has been announced as the 2023 recipient of the George M. Kober Medal from the Association of American Physicians. Congratulations!
https://t.co/5GLYvfQtu5
registration still open for upcoming workshop on eliciting structure in genomics data https://t.co/h2kYXVj70H Aug 30-Sept 3. Great line up of speakers. Come join us by zoom!