New vcfdist paper on bioRxiv!
Key Takeaways:
1) Jointly evaluating small and structural variants decreases measured FN+FP by 20-50%
2) 43-92% of phasing flip "errors" are FP due to variant representations
More below
Code: https://t.co/v3gz57lEPn
Paper: https://t.co/j9U7sQ4wCu
🚨Apparently all NIH Study Sections have been suspended indefinitely.
For those who don’t know, this means there won’t be any review of grants submitted to NIH
Depending on how long this goes on for, this could lead to an interruption in billions in research funding.
@AaronPomerantz@iiSeymour@rrwick@nanopore Cool! With 2/3 errors occurring in the same "GAGCTCC" motif, it makes me wonder if there's some kind of base mod, or if the motif is just underrepresented in the training data.
Climate experts were increasingly saying that keeping heating below 1.5C is near impossible, yet it remains the global goal. So I asked hundreds of top IPCC scientists what they thought. What they said shocked even me…
🧵 1/n #ClimateCrisis
I'm thrilled to announce that I successfully defended my PhD thesis today, "Improving select applications of long-read DNA sequencing"! After graduating, I'll be joining @fulcrumgenomics as a Senior Bioinformatics Scientist. I can't wait to see what this next chapter brings!
Mandatory reading and surprising results from Michael Hall and colleagues - much better SNP/indel calling results from latest nanopore (beating illumina), and deep learning methods really doing well. Extremely thorough work
https://t.co/Y4oQH1BeOc
Mandatory reading and surprising results from Michael Hall and colleagues - much better SNP/indel calling results from latest nanopore (beating illumina), and deep learning methods really doing well. Extremely thorough work
https://t.co/Y4oQH1BeOc
Excited to announce that our accompanying paper on HiPhase has been published today, I'll highlight some of the additions in this thread! #PacBio#HiFi
“HiPhase: Jointly phasing small, structural, and tandem repeat variants from HiFi sequencing”: https://t.co/sYFwXLlhcS
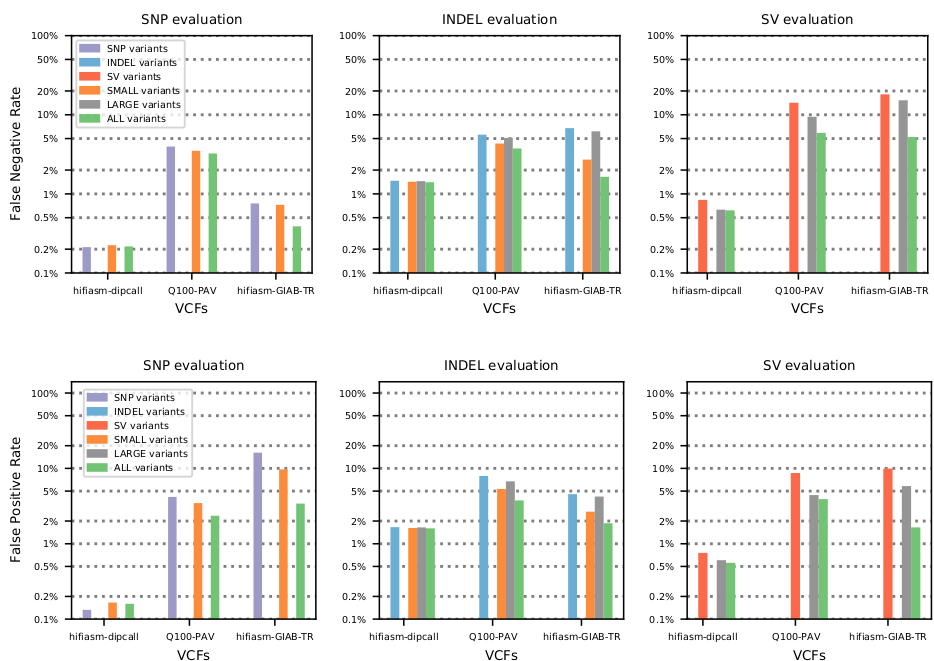
Comparison to prior work: lower measured FP and FN rates than vcfeval or Truvari, and similar results to Truvari refine (WFA/MAFFT). Truvari is faster and works with larger variants, but skips SNPs and the more accurate WFA/MAFFT versions don't scale to large BED regions
New vcfdist paper on bioRxiv!
Key Takeaways:
1) Jointly evaluating small and structural variants decreases measured FN+FP by 20-50%
2) 43-92% of phasing flip "errors" are FP due to variant representations
More below
Code: https://t.co/v3gz57lEPn
Paper: https://t.co/j9U7sQ4wCu
As noted above, joint evaluations reduce measured false negative and false positive variant calls across the board: by 28.1% for SNPs, 19.1% for INDELs, and 52.4% for SVs over 50 bases on 3 whole-genome datasets.