World models have made impressive progress in video generation, yet they still struggle with a fundamental challenge: memory. In long rollouts, the camera trajectory gradually drifts from the user-specified motion and revisited scenes no longer align with earlier observations. These errors accumulate over time, causing the generated world to steadily lose coherence.
🚀Excited to share our solution MosaicMem 🌍🧠 — our new hybrid spatial memory for video world models.
Project Page: https://t.co/nhy7nfGqLW
Paper: https://t.co/PpcMbq0JTY
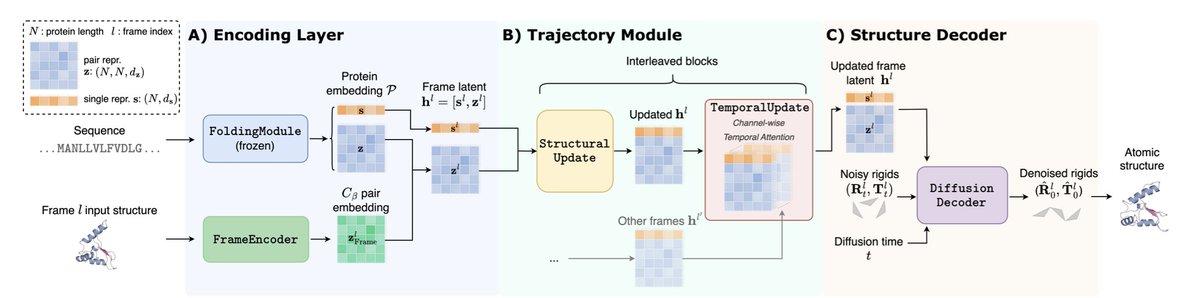
Excited to share STAR-MD: a scalable autoregressive diffusion model that generates stable, high-quality protein MD trajectories at microsecond timescales, where existing methods fail catastrophically.
Accepted at ICLR 2026! Links and details in the thread 🧵👇
1/7
Enjoyed the poster session at #NeurIPSanDiego with great conversations and insightful feedback!
ConfRover learns to generate high-quality protein conformation ensembles and temporal dynamics efficiently.
Model + inference code now released: https://t.co/vOvZCuMzzm
Simultaneous Modeling of Protein Conformation and Dynamics via Autoregression
1.This paper introduces CONFROVER, the first autoregressive model capable of simultaneously generating protein conformations and their dynamic trajectories, unifying time-dependent, time-independent, and interpolative sampling in a single framework.
2.CONFROVER models molecular dynamics trajectories through autoregressive factorization, allowing it to learn both conformational distributions and transitions directly from MD simulation data without relying on Markovian assumptions or fixed-length contexts.
3.Its modular architecture includes: (i) a protein-aware encoder leveraging OpenFold representations, (ii) a causal transformer-based temporal module for latent sequence modeling, and (iii) a SE(3) diffusion model for 3D conformation generation—ensuring high spatial fidelity and temporal flexibility.
4.Unlike prior models that treat conformations as discrete tokens or require separate models for different tasks, CONFROVER directly operates in continuous SE(3) space using denoising score matching loss, preserving geometric accuracy and enabling diverse sampling modes.
5.Experiments on the ATLAS dataset show CONFROVER significantly outperforms the previous SOTA MDGEN in trajectory simulation, with improved Pearson correlations in both RMSD and PCA space, indicating better capture of real protein dynamics.
6.It also matches or exceeds the performance of specialized models like ALPHAFLOW and CONFDIFF in time-independent conformation sampling, despite being trained as a general-purpose model—demonstrating strong versatility and generalization.
7.Through hybrid training (combining trajectory, single-frame, and interpolation objectives), CONFROVER can also generate smooth and accurate transition pathways between conformations, an essential feature for modeling functional state changes in proteins.
8.CONFROVER-MASK, a variant using masked sequence modeling, shows the framework's extensibility but lacks the inference flexibility and trajectory fidelity of the autoregressive (causal) version—highlighting the practical benefits of causal modeling for variable-length sequence generation.
9.In 100 ns simulation tests, CONFROVER achieves higher recall and F1 scores in recovering conformational states than MDGEN and shows strong alignment with principal dynamic modes extracted from MD using time-lagged ICA.
10.The model is efficient: generating an 80-frame trajectory (~100 ns) takes only 8.3 minutes, compared to 10 hours for traditional MD simulations—offering a substantial speedup for exploratory or large-scale applications.
11.While CONFROVER narrows the gap between deep generative models and physics-based MD, it still faces limitations in modeling large-scale conformational transitions and energy landscapes; further improvements may come from scaling data, adding energy terms, or optimizing structure modules.
12.CONFROVER provides a flexible and unified generative modeling framework for protein dynamics and structures, paving the way for more efficient, generalizable, and task-agnostic models in molecular simulation and protein engineering.
📜Paper: https://t.co/Zmz6xA97Kz
#ProteinDynamics #MolecularSimulation #AutoregressiveModel #SE3Diffusion #ProteinFolding #MDtrajectory #CONFROVER #ComputationalBiology #StructuralBioinformatics
CryoSTAR is officially published in @naturemethods! Huge thanks to our reviewers for their valuable feedback. In this final version, we show that an AlphaFold-predicted atomic model may serve as the reference, showcasing cryoSTAR’s flexibility. https://t.co/Aox8wq78kL
1/ 🔍 Wonder about the answer?
cryo-EM ✖️ Foundation Model 🟰 ❓
cryo-EM ✖️ Flow Matching 🟰 ❓
cryo-EM ✖️ Diffusion Transformer 🟰 ❓
Excited to introduce our new work--cryoFM, the first cryo-EM foundation model for protein densities with flow matching, which generalizes to four tasks without finetuning.
📃 Paper: https://t.co/YMyP2A50hu
🏠️ Project: https://t.co/O7OCzzduJF
Joint work with Yilai Li @li_yilai , Jing Yuan @eugenejyuan , Quanquan Gu @QuanquanGu
🚀 Our latest work on ProteinBench is a living benchmark for protein foundation models! 🧬🔬
The medal for #NobelPrize chemistry went to protein design and structure prediction, and ProteinBench is an ongoing "Olympics" in these two domains.
It now includes 20+ models and 9 key challenges, from designing functional proteins to generating protein dynamic structures. Checkout it for more details:
🤗Leaderboard: https://t.co/iCDbAmwFNY
📄 Paper: https://t.co/bJSMSGJfYp
🌐 Website: https://t.co/vV3EiJocMX
We welcome community feedback and participation to help this benchmark continue to grow!
@QuanquanGu@zaixiang_zheng@dngyxu1@YuningShen1@leowang_1
1/ 🎉 Exciting News! 🎉
Our paper "Protein Conformation Generation via Force-Guided SE(3) Diffusion Models" now has its corresponding code open-sourced! Dive into the details and explore the code at "https://t.co/bTO0Vabkuh" 🚀💻
@GoodNotesApp GN5 drained up my battery from 100% to 5% yesterday. It could last for 3~4 days when I was using GN4. Is there anyway I can export note from GN5 back to GN4? I have to stop using GN5 for now