A bit of news: After nearly 9 years, I have decided to leave Google DeepMind and join Anthropic (after taking some time to recharge). I am incredibly grateful for my time at GDM. @demishassabis took a real chance letting me lead the AlphaFold team just six months after finishing my PhD, and the entire GDM team taught me so much about how to do great science. GDM is a special place, and I’ll still be excited to hear about what amazing things they discover next.
We also found a scaling law that explains (p=0.03) Ab-Ag accuracy across model lineages - from Boltz to AF3 and beyond. 1.8x-ing AF3 performance (to match IsoDDE) would seem to require e.g. 10x more distillation and 16x compute. (7/8)
@mihirbafna14 and I are excited to introduce Promera, a co-folding and design model with
• best-in-class binder filtering
• nanobody design with in-silico success rates matching hallucination
• case studies on hantavirus epitope targeting and GPCR agonism (1/8)
Grok Build with Composer 2.5 visualizing predicted binders against PD-L1, a cancer immune-checkpoint protein that tumors use to switch off T-cells.
Composer 2.5 in Grok Build is using ChimeraX to visualize PD-L1 binders generated by Genie 3. In this workflow, a region on PD-L1 was selected for binding, Genie 3 generated binder shapes, ProteinMPNN filled in the amino acid sequences, and Boltz folded the binders against PD-L1.
Today marks my last day at ByteDance Seed.
Over the past 3 years, I had the opportunity to work across two of the most exciting frontiers in AI: AI for Drug Discovery and building frontier LLMs. Few opportunities in a career allow one to work simultaneously on curing disease and building frontier intelligence. I was fortunate to do both.
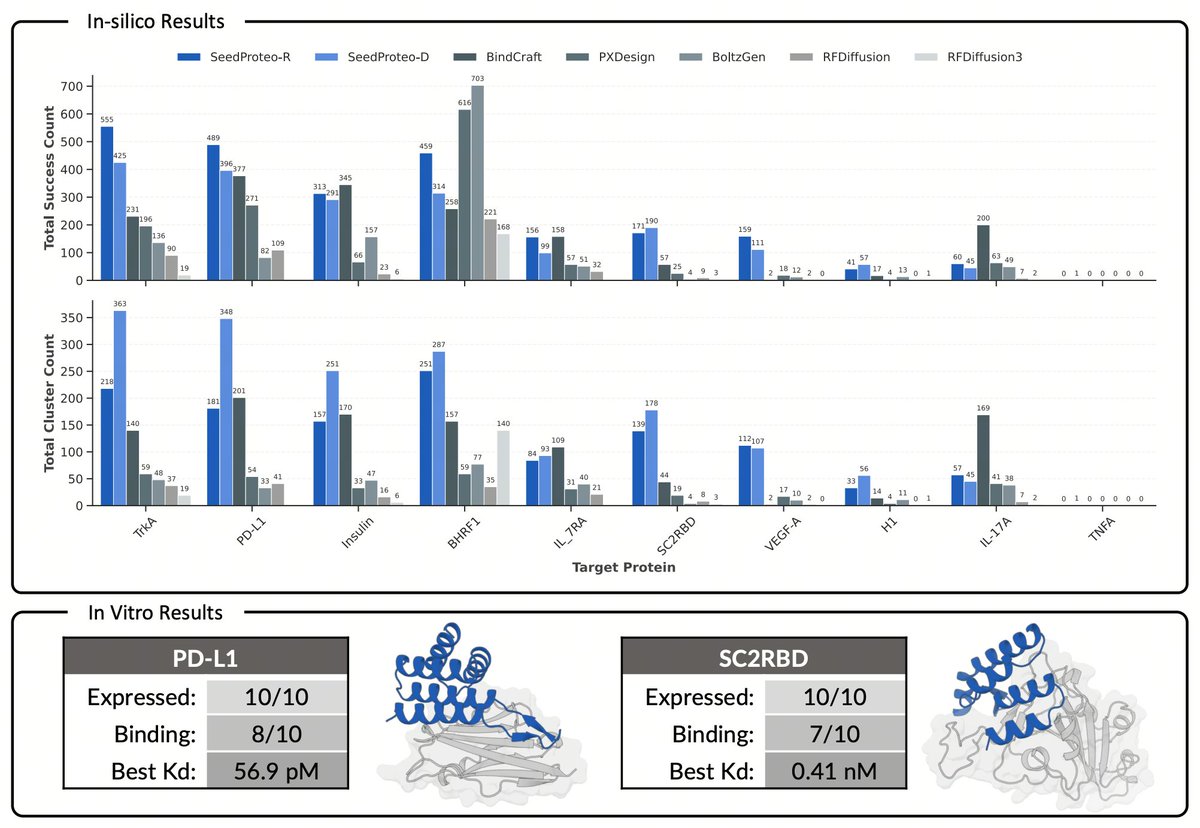
Since joining ByteDance, I have led the AI for drug discovery effort. Together with an exceptional team and collaborators, we built SeedFold, the world's first biomolecule structure prediction model to outperform AlphaFold 3 across a broad range of benchmarks and capabilities; SeedProteo, a state-of-the-art protein binder design model surpassing AlphaProteo, RFdiffusion, Chai-2, BinderCraft, and BoltzGen ; and the DPLM series of protein language models, along with several other ambitious AI for Science projects.
In early 2025, I took on a new challenge. To tackle one of the hardest problems in modern AI, reliably training and scaling frontier-scale LLMs, I joined the LLM pretraining effort and founded the LLM optimization and scaling team. Together, we built a highly scalable pretraining stack that enabled the successful training of Seed 2.0 and the subsequent frontier-scale models, significantly advancing our ability to train and iterate on frontier AI systems at scale.
I'm deeply grateful to my teammates, collaborators, and leadership for an incredibly rewarding journey.
The best model is yet to come.
Scaling continues!
Here is SeedProteo, our latest diffusion-based model for de novo all-atom protein design from ByteDance Seed! Our server is now live — feel free to give it a try! https://t.co/Tqsu2VPmWw
⚡️ A trend: prior from foundation models + likelihood from experimental data. 📖 Also suggest three related work: 1. CryoBoltz: Multiscale guidance of protein structure prediction with heterogeneous cryo-EM data (@ZhongingAlong ) 2. Inverse problems with experiment-guided AlphaFold (@AdvaithSai1 ) 3. CryoFM: A Flow-based Foundation Model for Cryo-EM Densities (from our team)
We introduce a new method, EmbedOpt, for robustly steering protein sequence-to-structure diffusion models to fit experimental data (Cryo-EM, NMR) without training. 🧬📉 @mhli41@JiequnH@PilarCossio2
EmbedOpt tackles the brittleness of the previous coordinate-space steering methods by optimizing the conditional embedding instead. These embeddings capture rich co-evolutionary signals in protein diffusion models—unlocking a new, robust and semantically meaningful diffusion steering axis.
🚀 Result: Better fitting, wider hyperparam stability, and efficiency enabled by fewer diffusion steps
📄 Preprint: https://t.co/Ir7QcXyIW9
The Iso team has cooked something incredible: our new technical report unveils the latest results from our drug design engine, the IsoDDE, progressing far beyond AlphaFold 3. This breaks new ground compared to AF and other similar methods by a significant degree across all key benchmarks. 1/7
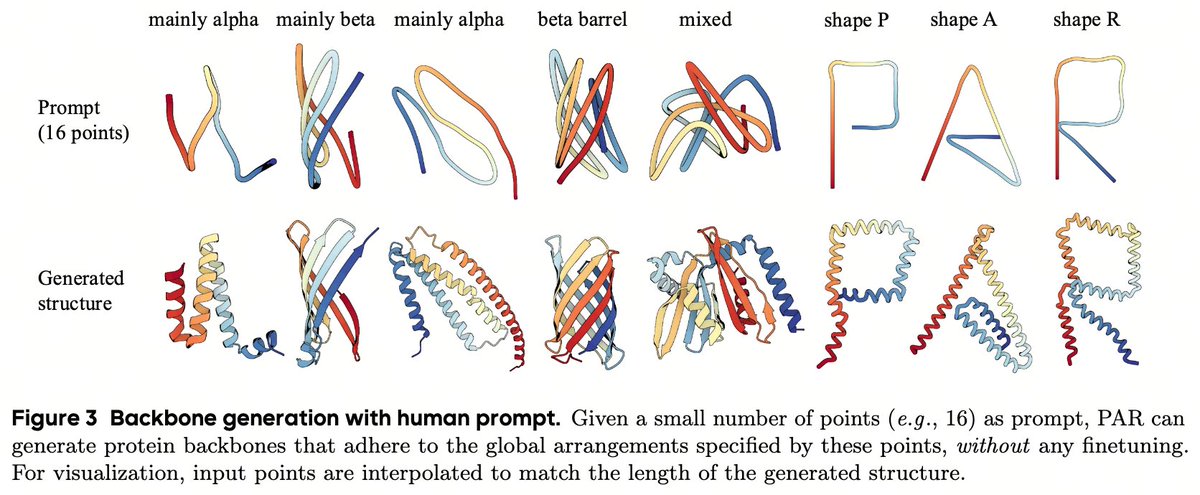
✍️ Zero-shot Prompted Generation:
Because PAR handles multiple granularities, a human can specify a coarse layout (e.g., just 16 points) and the model will generate a complete, structurally consistent protein backbone. No fine-tuning required!
Excited to share STAR-MD: a scalable autoregressive diffusion model that generates stable, high-quality protein MD trajectories at microsecond timescales, where existing methods fail catastrophically.
Accepted at ICLR 2026! Links and details in the thread 🧵👇
1/7
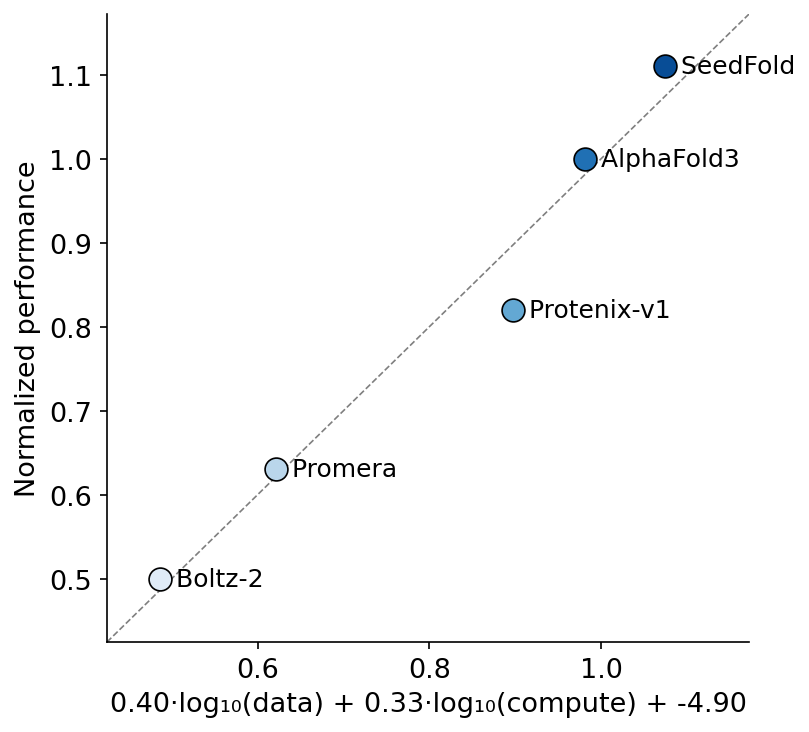
Tomorrow in the reading group we discuss "SeedFold: Scaling Biomolecular Structure Prediction" https://t.co/zKCQi5Z9dX
Outperforming AF3 on multiple tasks a little bit.
Join us on zoom at 9am PT / 12pm ET / 6pm CET: https://t.co/Kew3F4DXag
I think GEM has been the most interesting BioML workshop every year💙
- and I'm pretty pumped to attend hopefully submit and find this sort of workshop (eg. building the bridge to experimental biology) such a good fit for ML conferences b/c its not usual!