Flow matching is emerging as a unifying framework for generative biology
Biology is full of mappings between states: a healthy cell turning diseased, amino acids folding into a functional protein, a ligand docking into its target. Deriving such transformations analytically is intractable—which is where generative AI steps in, and flow matching is quickly becoming its backbone.
Morehead and coauthors review how flow matching (FM) is reshaping generative modeling in bioinformatics. Unlike diffusion models, FM doesn't force the source distribution to be Gaussian: it learns a time-dependent vector field that transports samples between any two distributions along straight-line, optimal-transport paths. The payoff: fewer inference steps, simulation-free training, and built-in support for geometric priors like SE(3) equivariance—essential for 3D biomolecules.
What's striking is how fast FM has spread across biological scales. For molecules, FoldFlow, FrameFlow, and Multiflow generate protein backbones on SE(3)ᴺ manifolds, SemlaFlow produces valid small molecules up to 100× faster than diffusion, and Dirichlet FM handles discrete DNA/RNA sequences. FlowDock and NeuralPLexer3 predict protein–ligand complexes that match or exceed AlphaFold 3 on key benchmarks, while AlphaFlow and MDGen generate conformational ensembles and MD trajectories. At the cellular scale, CellFlow and Meta FM map unperturbed populations to perturbed states, and CryoFM and FlowSDF extend FM to cryo-EM and microscopy.
The deeper point: FM subsumes diffusion models, continuous normalizing flows, and optimal transport as special cases, providing scaffolding for an AI-based virtual cell—simulating molecular, structural, and phenotypic effects of perturbations across scales.
Overall, this signals a shift in what's computationally tractable. Instead of narrow, stage-specific models, FM points to unified conditional generators that design sequences, predict complexes, and model perturbation responses in one framework—shortening wet-lab cycles and making closed-loop, active-learning workflows practical.
Paper: Morehead and coauthors, Nature Machine Intelligence (2026) — Journal license | https://t.co/7UyfTWXKmS
2. TIME: Tracking temporal effects is absolutely crucial. Tracking beginning & arbitrary end states only makes it impossible to trace the dynamics of cell state transitions, critical intermediate states, direct vs indirect effects, feedback & compensation etc. 5/
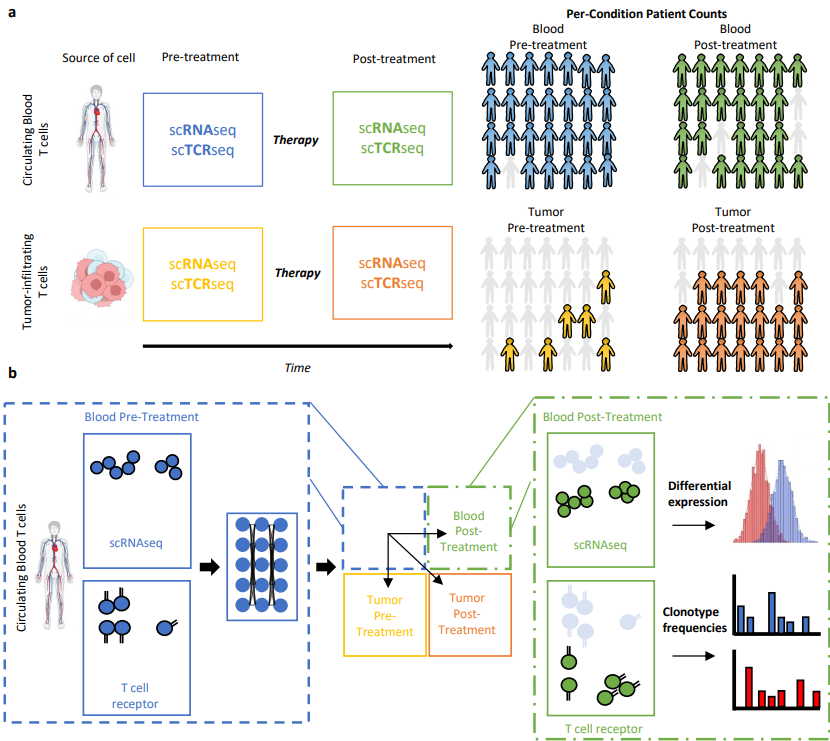
��� Congrats to @Schmidt_Center’s @ChujunHe123, @AmodioMatt, Orr Ashenberg, Caroline Uhler and colleagues on their Nature Communications paper! They introduce TRIM, integrating single-cell RNA + TCR data to study immune responses to cancer immunotherapy.
https://t.co/ZK7Dhm2Nbh
A non-hyped explainer of the “cell simulation” paper.
The recent study about the “4D” simulation of a minimal cell has been getting a lot of attention on social media. Unfortunately, most posts about it have serious errors. I’ve seen people claim that the model simulates every chemical reaction in the cell, for example, which is not true.
Some biomolecules and reactions *are* tracked individually in the simulation, including proteins and RNA (and ribosomes), and the chromosome. But the simulation does not track individual metabolites (like ATP or glucose), water, nucleotide precursors, lipds, and so on. These "other" molecules are represented, instead, as concentrations (using ordinary differential equations).
But anyway, here goes my quick explanation:
Researchers built a computational model that simulates roughly 100 minutes of biological time, or one cell division, for a single bacterial cell. Each simulation takes 4–6 days to run on two NVIDIA A100 GPUs, and the authors ran it 50 times in replicate. The cell simulation includes some elements of randomness, so each replication attempt leads to a slightly different outcome. When they plotted out these replicates and averaged results, they found that the model could predict a few things without being fitted to experimental data: The simulated cells “divided” every 105 minutes, on average, which matches experimental results; and the mRNA molecules had an average half-life of 3.63 minutes, which is roughly what we’d expect from experiments, too.
The cell they are modeling is called JCVI-syn3A, and it is not a naturally-occurring organism. It’s a bacterium that has been engineered, over many years, to have a small genome. It only has 493 genes (compared to 4,000+ for E. coli), all of which are housed on a single chromosome. The Syn3A cell was made by taking a natural organism, called Mycoplasma mycoides, and then slashing out non-essential genes. Its entire proteome, transcriptome, and metabolism have been studied in depth, which is why it’s being used to build these whole-cell simulations.
The actual *simulation*, though, is not a single thing! Instead, the authors wrote down all the “stuff” that happens inside a cell (transcription! translation! metabolism! lipid biosynthesis!) and decided which type of mathematical model would be best-suited to describe each thing. Some cell processes were modelled deterministically, others had “spatial” elements, and other parts were relatively random.
More specifically, they used four different types of models to build this simulation:
1. A Reaction-Diffusion Master Equation, which was used to model the individual proteins, RNAs, and ribosomes.
2. A Chemical Master Equation, which was used to model things where spatial location doesn’t matter as much (it basically treats the whole cell as one mixed entity); including tRNA charging.
3. Ordinary Differential Equations, which you may be familiar with from Calculus class, were used to model changes in ATP concentration, lipids, and so on.
4. Brownian Dynamics, which simulated the chromosome as a physical chain of beads, where each bead represents 10 base pairs of DNA.
The Reaction-Diffusion Equation works like this: Basically, they chopped up the entire digital cell into a 3D grid of cubes. Each cube measures 10 nanometers on each side. The whole cell is about 500 nanometers across, so there are tens of thousands of cubes in the cell's interior. (This is a useful way to coarse grain the simulation; if the cubes were smaller, the simulation would take much longer to run.)
Each cube is a little box that contains some number of molecules. At every “step” in the simulation, only one of two things can happen to the molecules in each box: Either they react with a molecule in the same box, or they diffuse (“hop”) to an adjacent box. That’s it; the model is just rolling a die for each molecule at each time step in each box, and using those results to decide how each molecule changes over time.
(The reason this spatial model is important is because biology only works if molecules physically bump into each other. And so this spatial grid means that, unlike simpler models, a protein actually has to “diffuse” across boxes in the cell to encounter its reaction partner; only then can it react and do something useful.)
So anyway, each of these models is used to represent a different type of molecule. It’s not like there is a single, all-powerful simulation that they are running here; instead, they’re running these four models together, using a script that synchronizes their results with each other.
The Reaction-Diffusion equation is the main part of the simulation. It takes time steps of 50 microseconds of biological time. Every 12.5 milliseconds of biological time — meaning every 250 RDME steps — the simulation pauses so that the other models can synchronize based on the latest state of the simulation. The Brownian Dynamics part runs on a completely separate GPU, and only updates every four seconds of biological time.
So that's the gist here. But let's also be honest about what this simulation does NOT do:
- It does not include polysomes, which are a cluster of ribosomes that all latch onto a single mRNA and translate at the same time. Polysomes are really common inside of cells, but this simulation assumes that each mRNA can only be translated by one ribosome at a time.
- It does not include polycistronic transcription. In bacteria, genes are often grouped next to each other on the chromosome and thus “transcribed” (or turned into mRNA) all at once, together. The majority of genes in E. coli, for example, are arranged in these operons, and the authors of this paper acknowledge that many Syn3A genes are likely co-transcribed the same way. But the simulation doesn't capture it.
- The authors manually tuned many parameters to get the model to make predictions that more closely resemble experiments. Earlier simulations were waaaayyyyy off from experimental results. For example, they adjusted the ratio of mRNA binding rates to ribosomes versus degradosomes because, in earlier simulations, mRNA was being degraded too quickly, before ribosomes could translate it, causing most proteins to be severely underproduced.
- In the Brownian Dynamics model, the authors added a “fake” 12 pN physical force to push the two daughter chromosomes apart during division, because the real biological mechanism for chromosome partitioning in Syn3A is not known.
- And some other things.
That being said: This model is really cool! I love papers like this! I'm enamored by scientists who choose really difficult problems (like simulating an entire cell) and actually go after it and make progress!
This paper is amazing because it shows us what we are able to simulate well, and what we don't yet understand, and to figure out which experiments we ought to perform to reconcile the two. So instead of framing this paper as "OH MY GOSH SCIENTISTS FIGURED OUT HOW TO SIMULATE AN ENTIRE CELL!" we should frame it as proof that there is still plenty of room at the bottom, many measurements to be made, and many avenues to explore as we seek to understand biology better.
Excited to share our new work. Over the past decade, single-cell genomics has transformed our ability to map cellular systems. But a major question remains:
Can we predict how perturbations reshape cellular trajectories over time?
In 2018, we first showed that it is possible to predict cellular responses to perturbations — ranging from disease signals to chemical treatments — even in unseen contexts. In 2022, we introduced CPA (MSB 2022; NeurIPS 2022), extending this idea to predict responses to unseen chemical and genetic perturbations, including their combinations.
Since then, the field of perturbation modeling has grown enormously. The community has pushed the space forward with many creative ideas and powerful models. It’s exciting to see how fast things are moving — even though many fundamental challenges remain.
One of the biggest is that cells are not static. They move through trajectories during development, immune responses, and disease. Yet most current models still predict perturbation effects within a single state, rather than how early perturbations propagate across future states and reshape downstream outcomes.
To address this, we developed PerturbGen, a trajectory-aware generative AI model that predicts how genetic perturbations reshape downstream cellular states.
Huge credit to the people who made this work possible. Thanks to co-first authors @lifeisscience_5, @Adib_m_, @Tomo_Isobe, @Amirhossein Vahidi, @delshadveghari & Anthony Rostron. Special recognition to @lifeisscience_5 and @Adib_m_ for driving this work over the finish line.
Grateful for our outstanding collaborators from @HaniffaLab, @BertieGottgens lab @GosiaTrynka and many others — a true cross-institute effort across @SCICambridge, @OpenTargets ,@sangerinstitute and @Cambridge_Uni.🎉
PerturbGen learns transcriptional dynamics across cellular trajectories. By introducing perturbations at an early source state, it can simulate how these effects propagate into future states along differentiation trajectories.
Scaling this across genes enables the creation of dynamic in silico perturbation atlases — maps of how perturbations reshape biological trajectories over time.
We explored this idea across three biological questions.
First, in a human in vivo LPS immune challenge, PerturbGen predicted that perturbing a transient IL1B signal dampens downstream inflammatory programs in myeloid cells, with pathway changes reversing signatures observed in an independent IL-1β stimulation experiment.
Second, in human hematopoiesis, PerturbGen predicted transcriptional responses to CRISPR transcription factor knockouts and enabled construction of perturbation atlases revealing lineage- and age-specific regulatory programs. These programs could also be linked to human genetics and blood diseases, including recapitulation of signatures associated with ETV6-related thrombocytopenia.
Finally, we asked whether perturbation modeling could help improve complex tissue models.
We built a dynamic perturbation atlas of human skin organoids to identify perturbations that could guideorganoid cells towardhuman fetal skin states.
PerturbGen prioritized activation of Wnt signaling via GSK3β inhibition. Experimental validation confirmed the prediction: treatment with CHIR99021 induced stromal gene programs and shifted organoid fibroblasts toward transcriptional states observed in fetal skin stroma.
Together, these results show how trajectory-aware perturbation modeling can connect gene perturbations to developmental programs, human genetics, disease mechanisms, and experimental interventions.
More broadly, we think these point toward a future where single-cell atlases become predictive systems.
As atlases expand across tissues, developmental windows, and modalities, models like PerturbGen could enable dynamic, virtual perturbation atlases— allowing us to simulate interventions, generate hypotheses, and design experiments before stepping into the lab.
Preprint
https://t.co/3peW7du2qM
Code
https://t.co/cmK0ymY5X7
Excited to see how the community builds on this work.
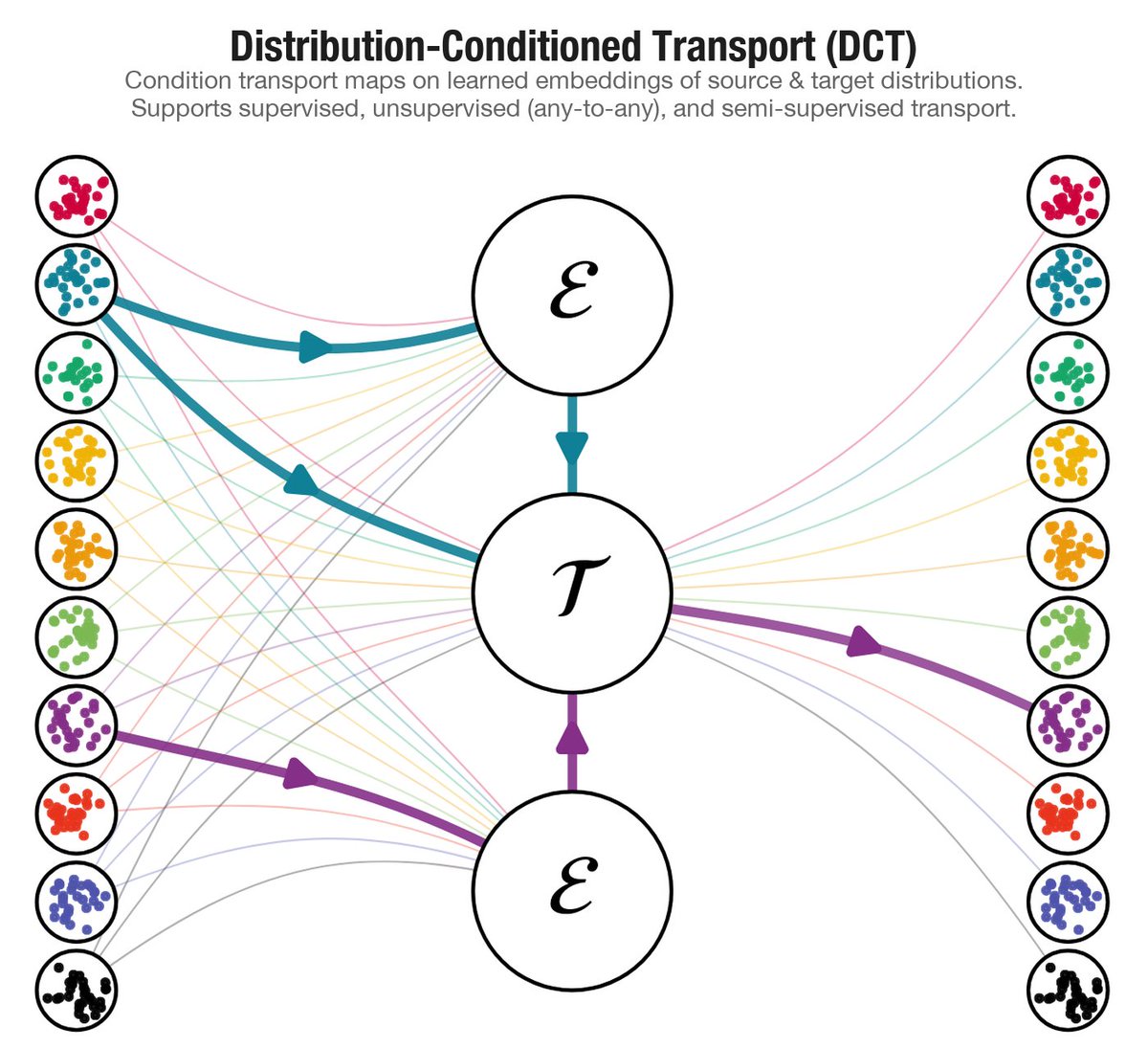
New paper: "Distribution-Conditioned Transport"
Modern scientific datasets don't contain one population — they contain thousands. Clones, donors, patients, each its own distribution. DCT learns transport maps that generalize across them, including to distributions never seen during training.
arxiv: https://t.co/vkTz5D2Gqq
Why I think your single-cell cell annotation benchmarking is missing the mark 👇
You trained your model on large of number of cells (millions), and you use your model to annotate a new dataset.
Huge thanks to my my mentors Natalie Stanley (@natstann, @comp_cy) & Justin Milner (@JJMilnerLab) and to my co-authors Genevieve Mullins, Will Green (@greenwilliamd), Huitong Shi, Haidong Yi, and Kay Chung (@HKayChung1) 🙏
10/
🕰️🚆🦠 Excited to share our new preprint: “Optimal transport fate mapping resolves T cell differentiation dynamics across tissues”!
We use OT to infer realistic CD8 T cell differentiation and migration trajectories from scRNA-seq timecourses.
https://t.co/rGzKrQV5qO
1/
We hope this illustrates how OT-based modeling paired with in vivo experiments can bridge computation and immunology–yielding actionable insights into T cell fate dynamics.
Would love to hear thoughts from those modeling single-cell dynamics, OT methods, or T cell biology.
9/