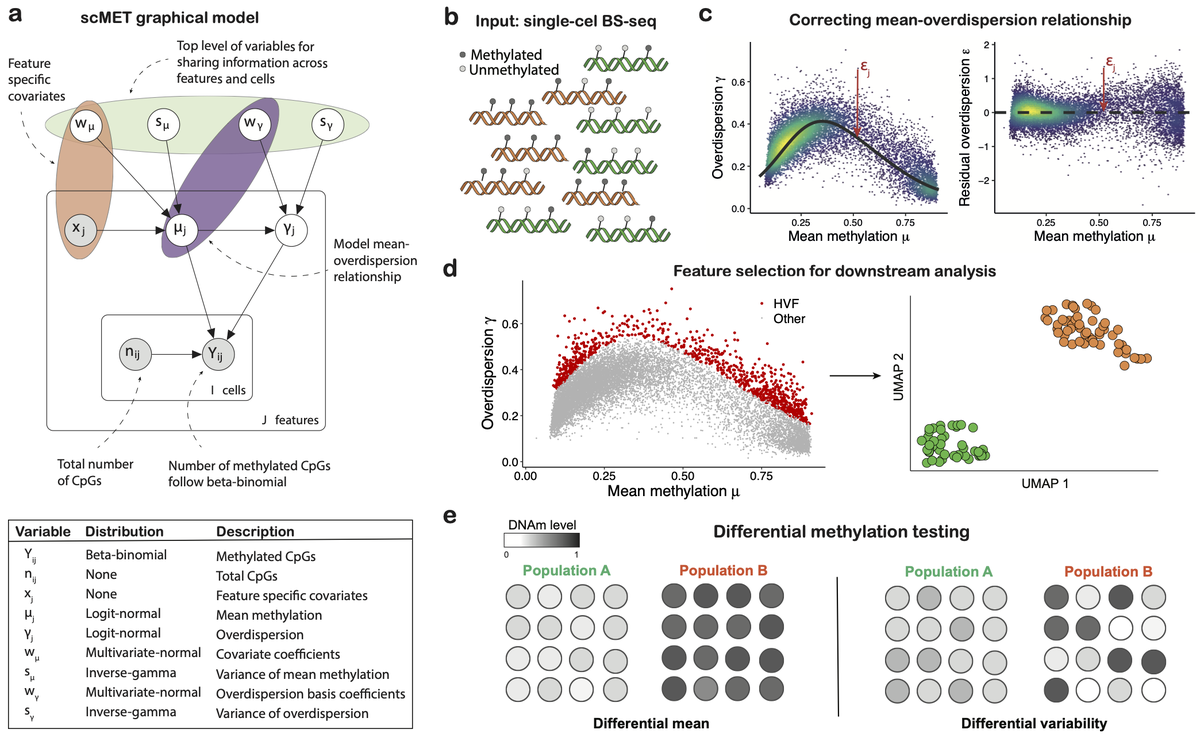
Really happy to share our latest preprint on quantifying cell-to-cell DNA methylation heterogeneity with scMET.
https://t.co/epM7coF7i4
Great team work with @RArgelaguet, Guido Sanguinetti and @CataVallejosM@mrc_hgu @MRC_IGMM @InfAtEd
Michael I. Jordan on the new MLST.
Four things:
> AGI is a PR term. It confuses young people.
> Discourse is bipolar, either alarmist or exuberant, this is in his words "so demoralizing" for 20- and 25-year-old researchers.
> ML's methods came from statistics and operations research, NOT the AI tradition.
> Data markets are Stackelberg games, not optimisation problems. A lot of ML researchers have never computed an equilibrium.
Michael I. Jordan is a no-nonsense original gangster of the field and was described by Science magazine, back in 2016 as the most influential living computer scientist.
Great to be at #ICLR2026 🇧🇷 presenting PatchDNA 🧬
If you’re around, come by our poster or reach out, always up for chatting about #ML for #Genomics, biology & drug discovery.
We’re also hiring @RelationRx across #ML/#AI, #CompBio & engineering.
https://t.co/GB7VKPwhsa
PatchDNA: A Flexible and Biologically Informed Alternative to Tokenization for DNA
1. PatchDNA introduces a novel approach to DNA language modeling by replacing traditional tokenization with dynamic patching, which segments DNA sequences into variable-length patches based on biological signals like evolutionary conservation. This method improves efficiency and allows models to focus on functionally relevant regions of the genome.
2. The key innovation is conservation-guided patching, which uses PhyloP scores to determine patch boundaries. This biologically informed strategy directs computational resources to conserved regions, which are more likely to be functionally important, leading to superior performance in various genomic tasks compared to existing tokenization methods.
3. PatchDNA also features re-patching, a capability that allows patching strategies to be modified after pretraining without retraining the model from scratch. This flexibility enables the model to adapt to different downstream tasks, such as cell-type-specific predictions, by incorporating context-specific signals like DNase-seq data.
4. In experiments, PatchDNA consistently outperformed or matched state-of-the-art models across multiple benchmarks, including regulatory element prediction, splicing, and gene expression tasks. Notably, it achieved strong results with significantly fewer parameters, demonstrating the efficiency of the patching approach.
5. The authors highlight that PatchDNA's framework supports various patching strategies, making it versatile for different biological applications. Future work could explore bidirectional encoding and multi-species data integration to further enhance its capabilities.
📜Paper: https://t.co/9evq2N719M
#DNALanguageModeling #Genomics #Bioinformatics #MachineLearning #ComputationalBiology
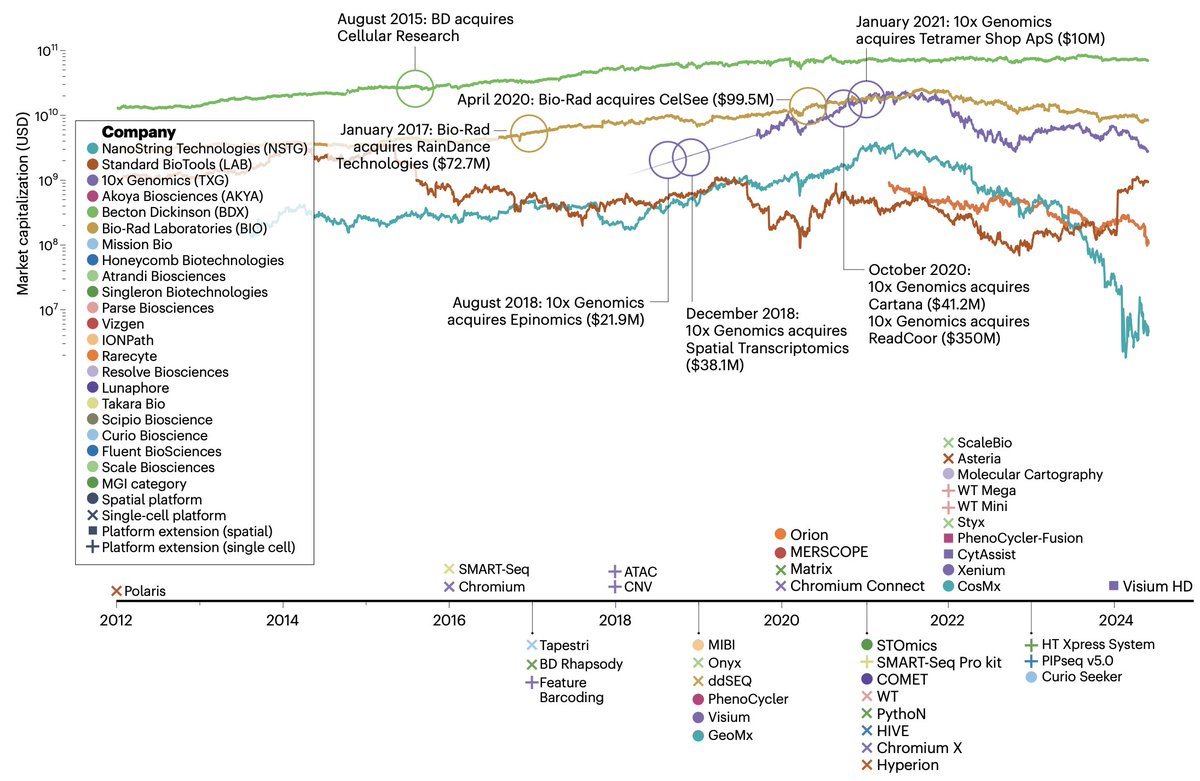
Today is the go live day for @scTrends_update! A consortium to both review *commercial* technologies, but also study market trends to assess angles for how technologies may differentiate themselves.
Have you ever wondered which single cell/spatial omic technology to use? There's lots of methods developed in academia, but no review of marketed products.
Check the @scTrends_update (https://t.co/aEMejm8Upm) commentary in @NatureBiotech by @wildtypehuman & @AdamCribbs
I am happy to share our new work on H3K9me3 in early embryos:
https://t.co/5jkFW2epnE
-H3K9me3 is inherited from the oocyte to the embryo (F1), like H3K27me3.
-HP1a binds independently of H3K9me2/3 in early embryos.
-The formation of HP1a foci is dependent on H3K9me2/3.
The self supervised single cell model race has clearly begun. Papers focus on architecture engg. boasting bigger models, bigger data, limited & often flawed benchmarks. Please move forward from benchmarking gym & show biological use cases & insights powered by these models.
I am delighted to share our recent paper: https://t.co/gP9Xvyi4iA.
It can be thought of as a version of the Bayesian Learning Rule, extended to the fully online setting.
This was a super fun project with @peterchang and the unstoppable Matt Jones (https://t.co/dgJtAkFRQC).
By leveraging 3 key ideas - natural gradient descent (way faster than OGD), implicit regularization to the prior (provably no need for a KL term!), and linearization (for fast and deterministic computation of expectations) - we get big speedups (both statistically and computationally) compared to online gradient descent and various Bayesian learning methods.
What happens when RNA polymerases encounters DNA damage during transcription? Excited to see our work out on quantifying the outcomes of such encounters in @PNASNews https://t.co/B1lAf3YpXo 1/n
https://t.co/A3GcqfDela
Here is an extended interactive visualisation. It makes it much easier to see why we get at most 6/8 regions no matter how we orient the 3 arrows.
In the late 1960s top airplane speeds were increasing dramatically. People assumed the trend would continue. Pan Am was pre-booking flights to the moon. But it turned out the trend was about to fall off a cliff.
I think it's the same thing with AI scaling — it's going to run out; the question is when. I think more likely than not, it already has.
So much happening in ML for Biology and Health at ICLR & MLDD this year and so little time to catch up?
We've got you covered with a quick summary of #ICLR2024 and MLDD content focused at the buzzing intersection of Bio, Health and AI!
thread👇
I want to explain a statistical mechanical concept known as coarse graining which I think might be useful for thinking about things like AF3. Especially a special case known as continuum or implicit solvent models.
Our paper (Mixed Models for Multiple Instance Learning) got an ORAL and OUTSTANDING STUDENT PAPER at AISTATS 2024 🎉💯🎉🚀
📃: https://t.co/wQUpKnvza4
💻: https://t.co/pajra4aZYe
Congrats to @janxengelmann@ale__palmaa@fabian_theis@fpcasale 👏 Hard work pays off 🔥
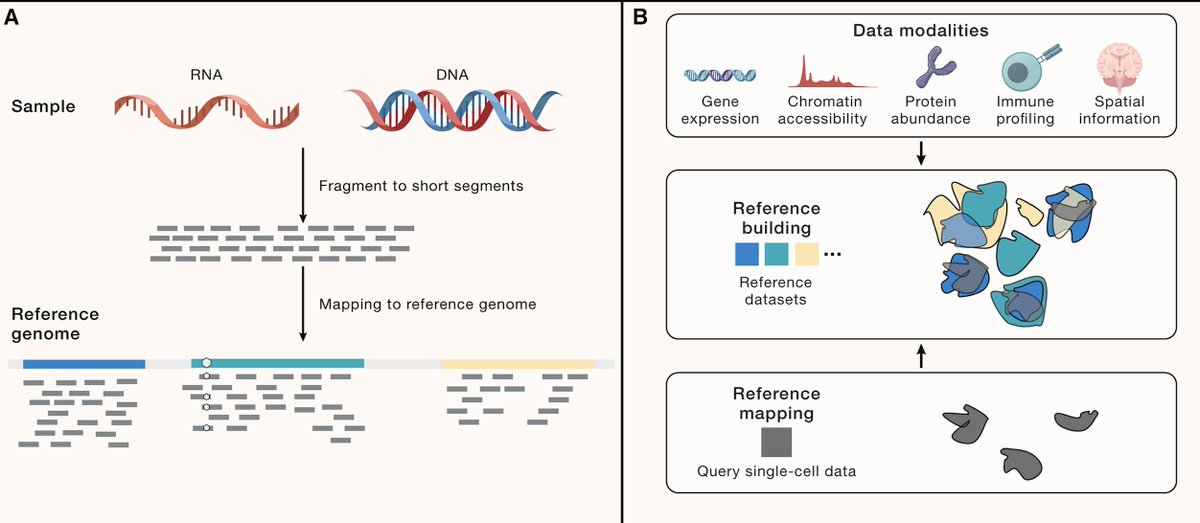
Computational methods have enabled the creation of large-scale single-cell atlases, akin to reference genomes, but these are costly and complex to build. Our perspective at @CellCellPress, alongside @YUHANHAO2, @satijalab, and @fabian_theis, discusses how the reference mapping paradigm maps new data onto existing atlases to integrate datasets across health states, perturbations, modalities, and species. This approach promises to automate single-cell data analysis, reducing reliance on manual, labor-intensive processes.
read here: https://t.co/QDV7Ah5Lcd
Could not be more delighted to present our work investigating how over 220,000 complex and molecular trait-associated genetic variants affect transcriptional regulation using massively parallel reporter assays!
https://t.co/1eoN4OxAvd
See below for a 🧵. 1/n
A bit slow on X but still super excited to share our Hist2Cell, predicting fine-grained cell types (e.g., 80 subtypes in lung) solely from histology images. For less funded labs to go for spatial RNAs, you can observe a lot more just from H&E stains (1/4) https://t.co/Q48GqrUeD7