Assistant Professor @KAIST Grad School of Medical Science and Engineering, P-Chem PhD @Caltech, postdoc Long Cai lab @Caltech and Kwanghun Chung lab @MIT.
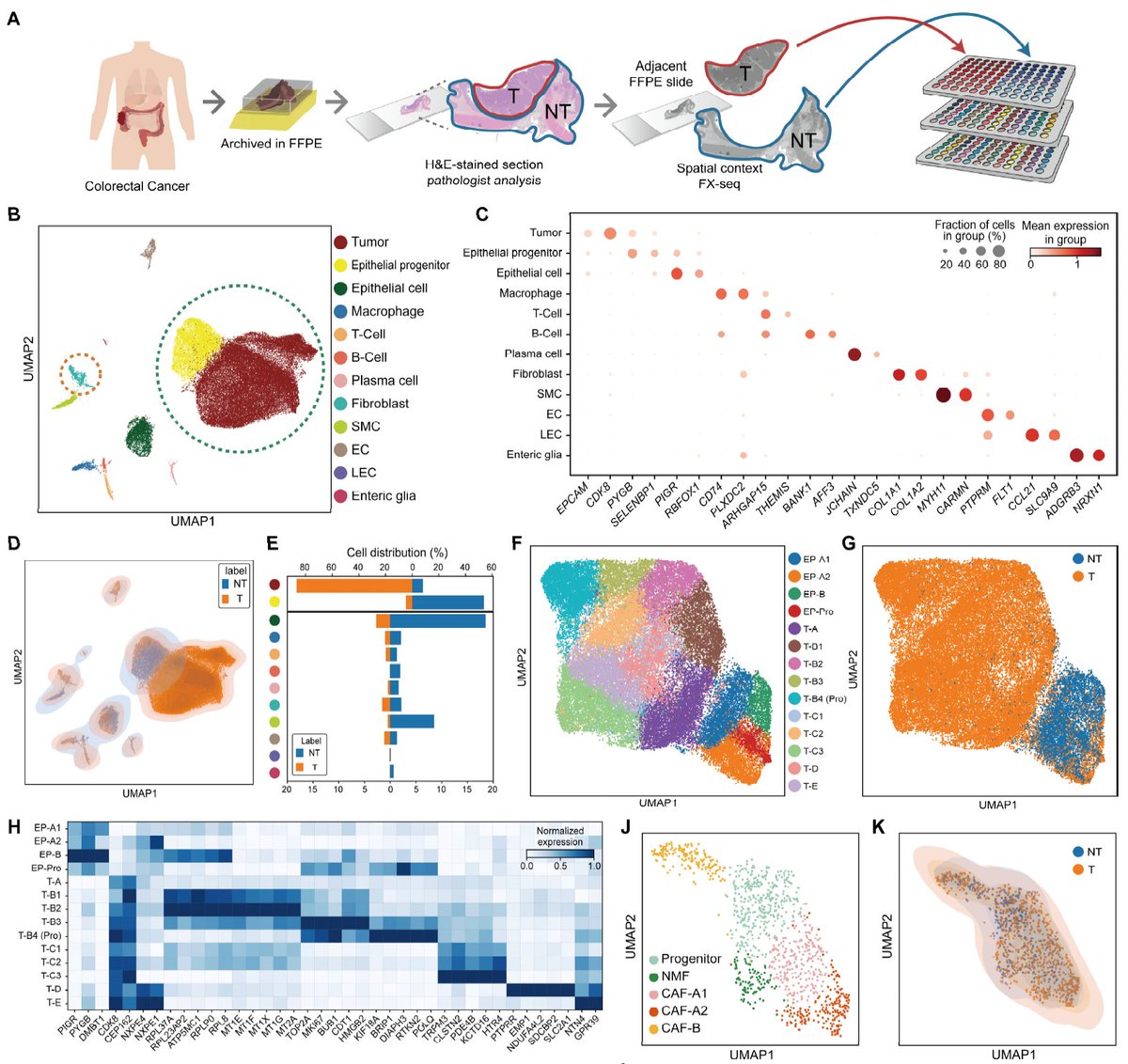
🚨 Introducing FX-seq (Fixative eXchange-Seq)! We are thrilled to share our preprint on bioRxiv. We've developed a robust method to unlock high-quality single-cell transcriptomes from heavily fixed FFPE clinical tissues. 🧵👇
https://t.co/97eqyoE4MN
New preprint alert! 🚀 Across 60 human cancer cell lines spanning 20 cancer types, single-cell RNA+ATAC reveals pan-cancer cell-state heterogeneity, core gene-regulatory networks, and an EMT axis conserved across tissue origins.
Huge congrats to first author @xu_zxu, who recently defended his PhD, and Aileen Ugurbil from our lab @RockefellerUniv! https://t.co/4fNQJlJY3m
New preprint alert🚀 Using TrackerSci, we quantified the birth rate of each cell type across the whole organism, and how these rates are rewired with age.
Huge congrats to the incredible Dr. Lu @ziyu__lu from @RockefellerUniv, who just defended today!
https://t.co/h50Swm03Lh
1/ Thrilled to share our new paper, out today in @ScienceMagazine! We built a pan-cancer spatial atlas of tertiary lymphoid structures (TLSs) and developed computation and AI frameworks to study TLS biology at scale.
https://t.co/zgcBnnm7Rl
Today we're announcing ESMFold2, an open scientific engine to power prediction, design, and discovery across protein biology.
The new model delivers state of the art performance on protein interactions, especially antibodies, a critical modality for therapeutics.
We have designed and validated miniprotein binders and single chain antibodies across five therapeutic targets that are important in cancer and immunology. We are seeing very high success rates, and affinities at levels consistent with therapeutic activity.
We’re also releasing an atlas of 6.8 billion proteins, and 1.1 billion predicted structures.
ESMFold2 is built on a state of the art language model that has been trained on billions of protein sequences.
A world model of protein biology emerges through language modeling.
We’ve used the techniques of mechanistic interpretability developed to understand large language models to understand the concepts ESM uses to represent proteins.
The model’s representation space has a compositional organization of features across scales, levels of complexity, and abstraction, that reflects and mirrors the understanding of protein biology developed through a century of empirical science.
This understanding emerges without prior knowledge, just from language modeling of protein sequences.
Language models are becoming a powerful substrate to understand and program biology.
The design of protein interactions is one of the most fundamental problems in biophysics, and has critical implications for the discovery of new medicines. A simple gradient based search with the model was able to discover high-affinity protein binders.
I'm excited by the potential this has to accelerate basic science and the understanding of proteins. And especially for the new avenues it opens up for therapeutic design and medicine.
AI can now design antibodies that bind with atomic precision, but not ones that cells can produce. Our preprint closes this gap, delivering a structural principle, an AI-guided rescue pipeline, and adalimumab variants with 20-100x in vivo potency.
https://t.co/GvfgHA5EcU
🚀Excited to share our new paper @Nature! We developed PerturbFate platform to discover how diverse genetic perturbations converge on a shared cell state, and key molecular programs driving it, led by our incredible student @xu_zxu from @RockefellerUniv.
https://t.co/NLHUVIB6Dg
The AI Scientist: Towards Fully Automated AI Research, Now Published in Nature
Nature: https://t.co/nNfpSV5e5I
Blog: https://t.co/i6h8LVQOdl
When we first introduced The AI Scientist, we shared an ambitious vision of an agent powered by foundation models capable of executing the entire machine learning research lifecycle.
From inventing ideas and writing code to executing experiments and drafting the manuscript, the system demonstrated that end-to-end automation of the scientific process is possible.
Soon after, we shared a historic update: the improved AI Scientist-v2 produced the first fully AI-generated paper to pass a rigorous human peer-review process.
Today, we are happy to announce that “The AI Scientist: Towards Fully Automated AI Research,” our paper describing all of this work, along with fresh new insights, has been published in @Nature!
This Nature publication consolidates these milestones and details the underlying foundation model orchestration. It also introduces our Automated Reviewer, which matches human review judgments and actually exceeds standard inter-human agreement.
Crucially, by using this reviewer to grade papers generated by different foundation models, we discovered a clear scaling law of science. As the underlying foundation models improve, the quality of the generated scientific papers increases correspondingly. This implies that as compute costs decrease and model capabilities continue to exponentially increase, future versions of The AI Scientist will be substantially more capable.
Building upon our previous open-source releases (https://t.co/H1tBT14Yx8), this open-access Nature publication comprehensively details our system's architecture, outlines several new scaling results, and discusses the promise and challenges of AI-generated science.
This substantial milestone is the result of a close and fruitful collaboration between researchers at Sakana AI, the University of British Columbia (UBC) and the Vector Institute, and the University of Oxford. Congrats to the team!
@_chris_lu_@cong_ml@RobertTLange@_yutaroyamada@shengranhu@j_foerst@hardmaru@jeffclune
🚨 Introducing FX-seq (Fixative eXchange-Seq)! We are thrilled to share our preprint on bioRxiv. We've developed a robust method to unlock high-quality single-cell transcriptomes from heavily fixed FFPE clinical tissues. 🧵👇
https://t.co/97eqyoE4MN
Their creativity, persistence, and scientific leadership were absolutely critical throughout this long and challenging journey. I’m truly thankful for what they’ve built and how far they’ve pushed this technology.
I am deeply grateful to the brilliant team who made this possible. Massive shoutout to Han-Eol Park and Yu Tak Lee, who played the central roles in conceptualizing, building, and advancing FX-seq. 👏
We see FX-seq as a foundational step toward FFPE-native single-cell genomics, pathology-linked spatial biology, and next-generation biomarker discovery from the samples that matter most in the clinic.
And there is much more to come. We are currently finalizing our revisions and will soon share expanded, highly adaptable protocols compatible with major commercialized platforms. Stay tuned!
The result is a highly robust and reproducible workflow. In this initial preprint, we demonstrate its power and validate the core biochemistry using a custom sciRNAseq3-based cell barcoding approach.
How does it work? We systematically optimized the entire process to dramatically improve reverse transcription yields, finally enabling highly efficient 3' priming for FFPE samples.
Unlike conventional fresh-tissue-centered workflows, FX-seq is built from the ground up with real-world clinical samples in mind. We believe FFPE tissues should be a first-class resource for genomics, not just leftovers from pathology archives.
Enter FX-seq. Our goal was to unlock the vast transcriptomic information trapped in heavily fixed clinical tissues, making these invaluable archived specimens accessible for single-cell and spatial analysis.
Most human disease biology in the clinic is stored in FFPE tissue blocks, yet modern single-cell & spatial genomics have largely been built for fresh samples. Bridging this critical gap has been a major unmet need in translational medicine.