🚀 New tool out! LIVIA (Local Interaction Visualization and Analysis) — a browser-based tool for assessing and visualizing predicted protein-protein interactions.
Drop in a prediction from AlphaFold-Multimer, AF3, ColabFold, Boltz-1/2, Chai-1, or OpenFold3 (ZIP or folder, auto-detected) and LIVIA answers the two questions you actually care about:
▸ Do these proteins interact?
▸ Which residues form the interface?
What you get:
▸ Interface confidence scores — iLIS (our local metric), ipSAE, actifpTM, ipTM
▸ Interaction interface heatmaps (PAE, LIS, cLIS)
▸ Sequence viewer + linear & circular contact maps highlighting Local Interaction Residues (LIR) and contact LIR (cLIR)
▸ Embedded Mol* 3D viewer
▸ Downloadable ChimeraX & PyMOL scripts
Also fetches dimers directly from the AlphaFold Database — adding the interface annotations AFDB doesn't provide.
Everything runs locally in your browser. No install, no upload.
LIVIA started as a personal tool — I built it with Claude Code and used it to make every structure figure in our FlyPredictome preprint (Kim et al., 2026). (Claude Code is truly insane...) Along the way I realized it could be useful for others, too.
If you have a favorite color palette for structure visualization, please let me know — happy to add it as a preset 🎨
🔗 LIVIA tool: https://t.co/qSaedW3yka
🐍 iLIS / batch CLI: https://t.co/jYN3yg3l6a
📄 LIVIA preprint: https://t.co/uHwEbAMBGH
📄 FlyPredictome preprint: https://t.co/O3mAOzF2GD
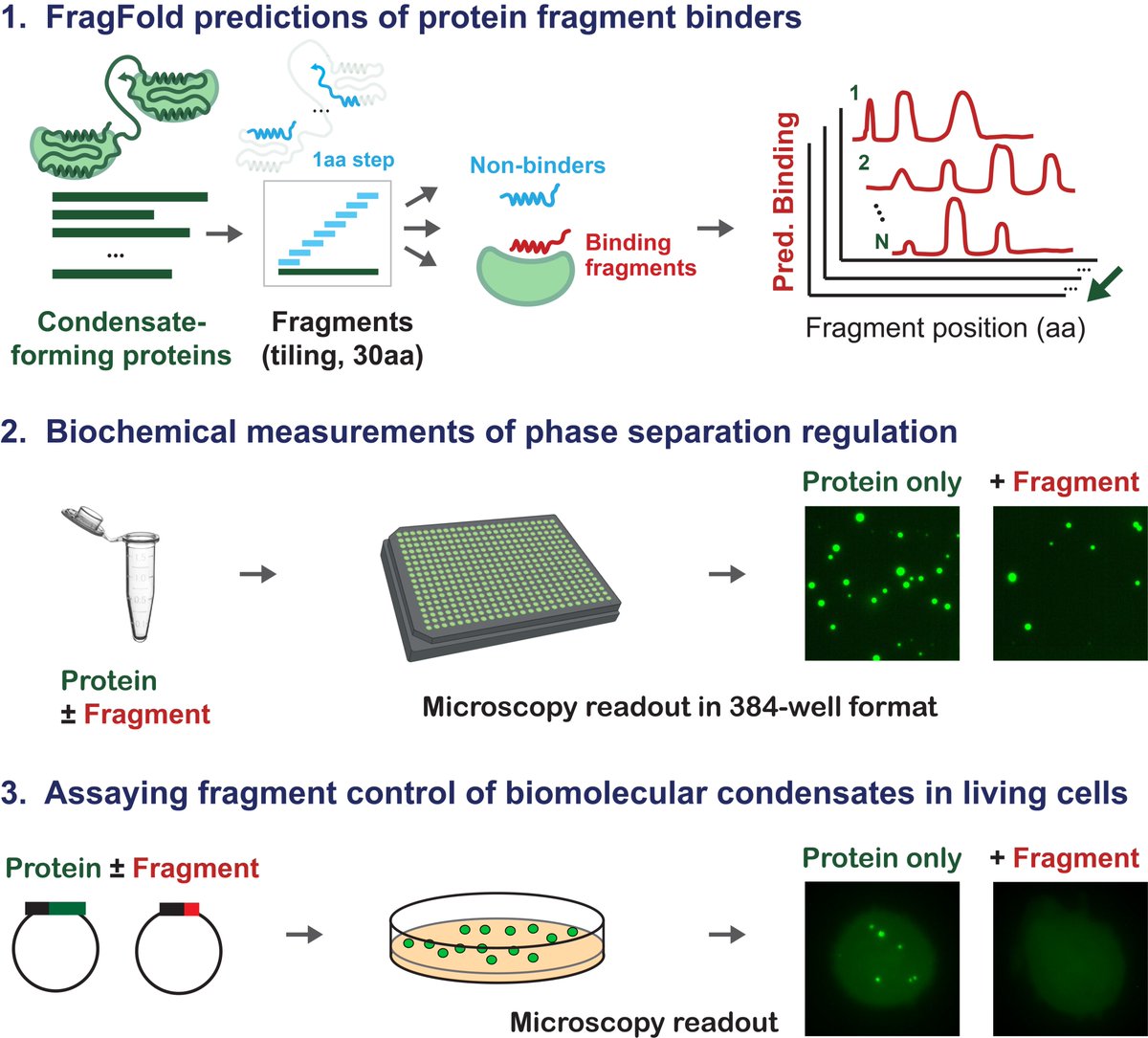
Interested in genetically encodable inhibitors of your favorite biomolecular condensate? Excited to announce our latest work, w/ @jibin_sadasivan, @GeneWeiLiLab, & @LindsayCase19, on protein fragments as generalizable regulators of phase separation. (1/n)
https://t.co/6IMkrTP3ZQ
We just made an app that walks you through designing a novel protein with AI from scratch. Takes about 5 minutes, requires zero biology knowledge.
➡️ https://t.co/L3dg6H6BTU
The best part: we will actually synthesize 1000 of those protein designs in the lab and test their real world function as a therapeutic.
Excited to share our review: “Twenty-Five Years of Photobodies: Formation, Composition, and Two-Compartment Phytochrome B Signaling Logic.” We synthesize how photobodies form, what’s inside, and how PB-nucleoplasm partitioning tunes phyB signaling dynamic range. https://t.co/jiJI036DYd
👩🔬🧑🔬 La Junta convoca ayudas por valor de 9,01 M€ para contratar a 65 investigadores doctores, por un periodo de 3 años y bajo el compromiso de crear una plaza con el mismo perfil de la ayuda concedida.
🗓️ El plazo abre el próximo 5 de enero de 2026.
🔗 https://t.co/araAgHr811
Here's how to predict binding of tens or hundreds of small molecules to protein assemblies using Boltz 2 in ChimeraX. Accuracy depends on how similar the ligands and binding pockets are to existing experimental structures. https://t.co/6lRBlT31o4
Diffusing Protein Binders to Intrinsically Disordered Proteins
🚀 New paper from David Baker!🚀
1. A groundbreaking study published in Nature demonstrates a novel method for designing protein binders that can target intrinsically disordered proteins (IDPs) and regions (IDRs) with high affinity and specificity. This approach leverages RFdiffusion, a powerful computational tool that generates binders by freely sampling both target and binding protein conformations from the target sequence alone.
2. The research team successfully created binders for a diverse set of IDPs and IDRs, including amylin, C-peptide, VP48, and BRCA1_ARATH, with dissociation constants (Kd) ranging from 3 to 100 nM. These binders were shown to bind their respective targets in cells, highlighting the potential for therapeutic and diagnostic applications.
3. A key innovation is the ability to design binders without pre-specifying the target geometry, allowing the binder to select a specific conformation from a broad ensemble of possible states. This induced-fit mechanism is particularly effective for IDPs, which lack a single well-defined structure.
4. The study also introduced a two-sided partial diffusion approach to optimize binding affinity, resulting in binders with significantly improved metrics compared to traditional one-sided diffusion methods. This technique allows both the target and binder to adapt their conformations during the design process.
5. For shorter IDRs, the researchers incorporated secondary structure specification into the RFdiffusion model, enabling the design of binders that interact with β-strand conformations of the target. This method significantly increased the efficiency of generating high-affinity binders.
6. The designed binders were validated through crystallography, showing close agreement between the computational models and experimental structures. Additionally, the binders demonstrated high specificity for their intended targets in all-by-all binding experiments.
7. In cellular experiments, the binders colocalized with their full-length targets, confirming their ability to engage targets in a cellular context. Notably, the G3BP1 binder disrupted stress granule formation, while the amylin binder inhibited amyloid fibril formation and dissociated existing fibers.
8. The amylin binder was further shown to enhance the sensitivity of mass spectrometry-based amylin detection and enable lysosomal targeting of amylin monomers and fibrils, demonstrating the potential for improving diagnostic techniques and therapeutic interventions.
9. This work represents a significant advancement in the field of protein design, offering a versatile and powerful approach to targeting IDPs and IDRs. The method has broad implications for understanding and modulating protein interactions in various biological contexts.
💻Code: https://t.co/EsmkpqRMiJ
📜Paper: https://t.co/KlFoXcwsSR
#ProteinDesign #IDPs #Therapeutics #ComputationalBiology #Innovation
More than 2700 3′UTRs are highly conserved.
These 3′UTRs are essential components in mRNA templates, as their deletion decreases protein activity without changing protein abundance.
Highly conserved 3′UTRs help the folding of proteins with long IDRs.
https://t.co/b6hd4AlIoX