RFdiffusion3 now available! De novo protein design against any molecule
Try it on @tamarindbio today
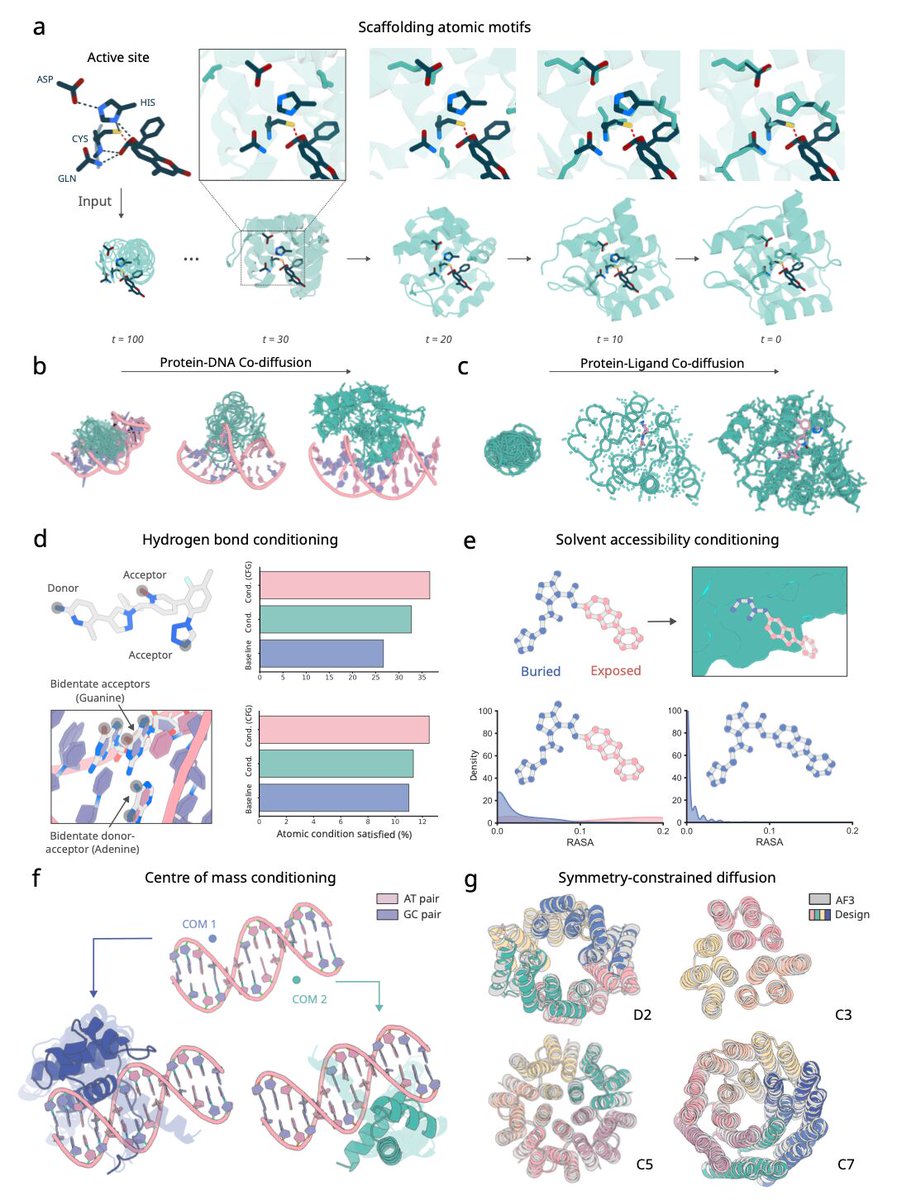
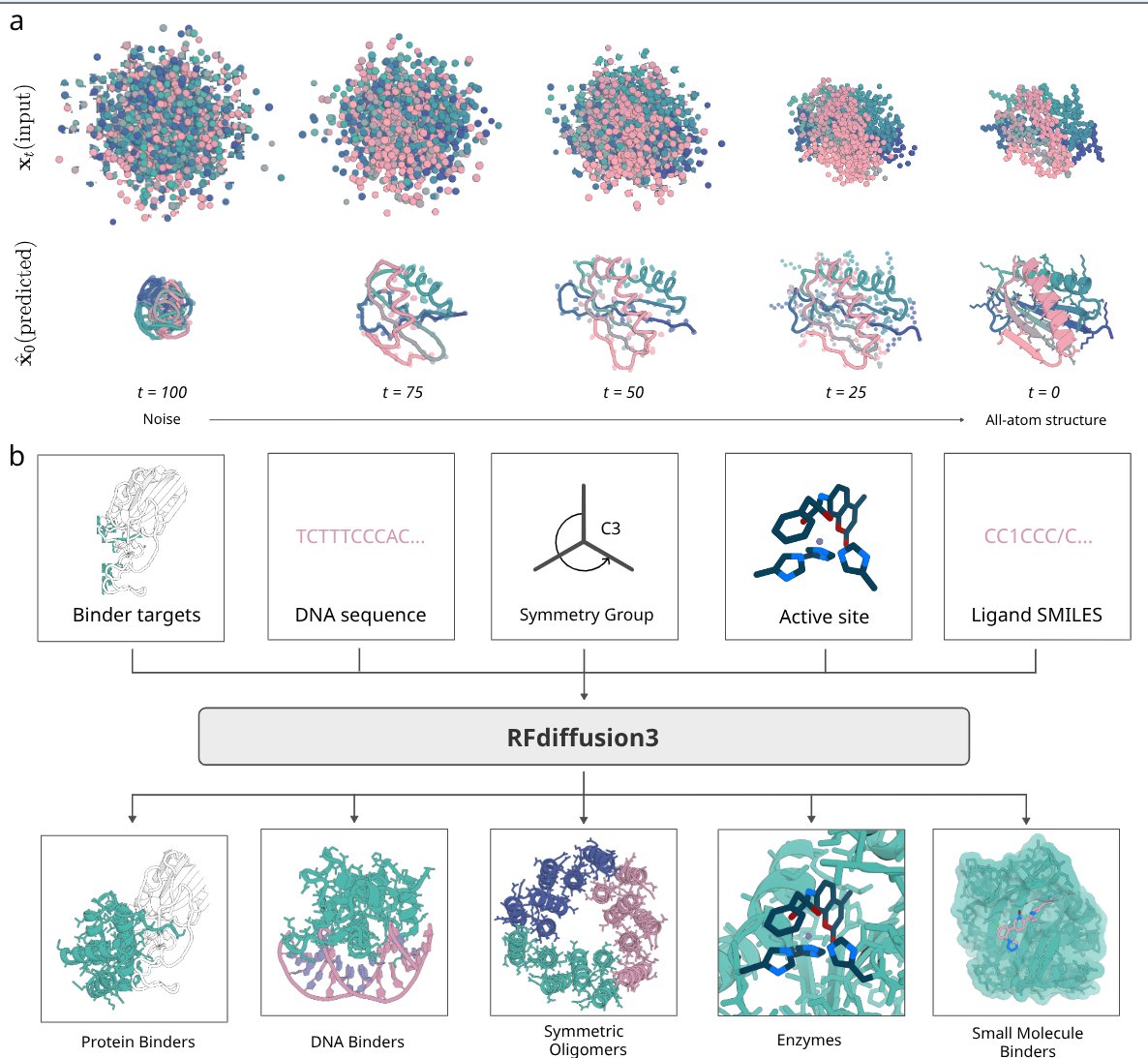
RF3 shows success in designing de novo proteins against all-atom targets, including proteins, DNA and small molecules with diverse applications.
Today, we report a method for design of active enzymes, RFdiffusion2, in @naturemethods. For the first time, we are able to design enzymes with native-range catalytic activity.
We also are releasing our next frontier model, RFdiffusion3, code 👇
Excited to share Caliby 🐈, our new model for structure-conditioned sequence design! Caliby is a Potts model-based sequence design method that can condition on structural ensembles. We use this to average out non-structural signal (e.g. evolutionary bias) learned by models 🧵1/N
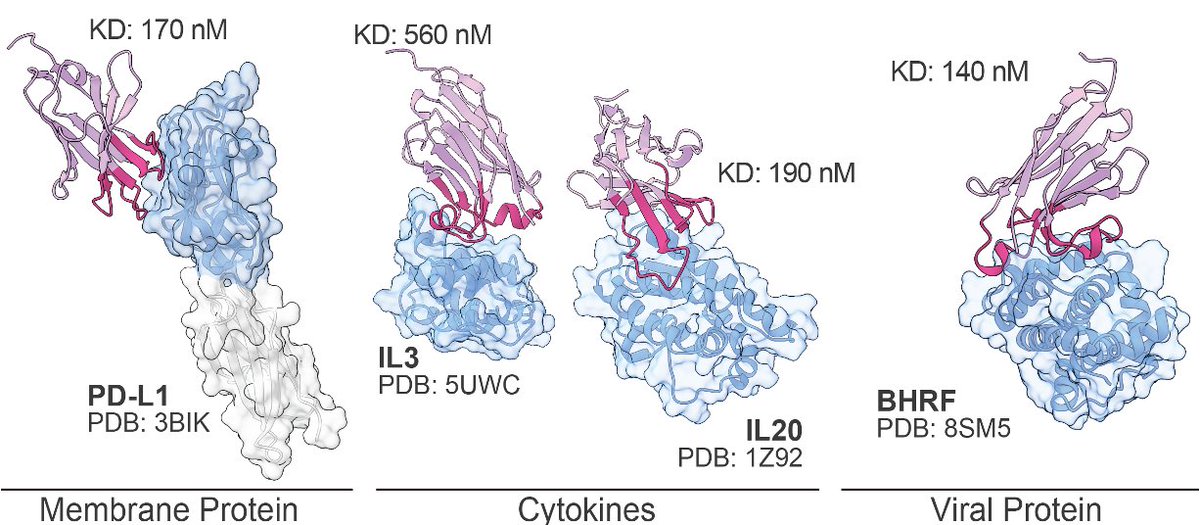
The ability to design antibodies against any protein of interest has major implications for medicine, biotech, and basic science.
Today, we introduce Germinal, a pipeline for epitope-targeted de novo antibody design achieving 4–22% success rates with efficient experimental validation.
✨ ReaSyn ✨
A generative model designed for predicting molecular synthesis pathways. ReaSyn reframes molecule synthesizability as a chain‑of‑thought (CoT) problem.
ReaSyn’s step‑wise ‘reasoning’ generates designable molecules, crushing prior SOTA in reconstruction & hit expansion.
Will CoT‑style reasoning become chem‑tech’s new default?
Explore the paper, technical blog, and code 🧵
We asked ourselves what could we do if we worked cooperatively as a team to create a single model that could design any biomolecular interactions.
Yesterday, we reported details of RFdiffusion3, the next step towards building functional proteins straight from the computer.
Great to finally have this available for people 🙌
Thank you to everyone involved, especially @nscorley & @r_krishna3! We'll be pushing a few smaller updates in the coming days to enhance usability for everyone (including some docs on what you can do with it) - so stay tuned (:
RFDiffusion3 generates all atom bound conformation, making it significant for flexible targets like DNA.
An excellent teamwork to achieve something impossible by any one of us in just few months.
@butcher_jasper@r_krishna3
https://t.co/i60oujJS1L
Accelerating Biomolecular Modeling with AtomWorks and RF3
🚀 New preprint from David Baker!🚀
1. A new framework called AtomWorks has been introduced to revolutionize biomolecular modeling. AtomWorks provides a unified and modular platform for developing state-of-the-art biomolecular models, including structure prediction, protein design, and sequence design. It streamlines the process of data preparation and model training, making it easier for researchers to prototype and test new ideas.
2. The AtomWorks framework emphasizes high-quality data handling. It standardizes inputs from diverse sources, such as the Protein Data Bank (PDB), and resolves common issues like incorrect bond orders, charges, and missing coordinates. This results in higher-quality derived features and improved model performance. For example, AtomWorks-generated reference conformers have lower energies compared to those from other open-source models.
3. AtomWorks enables rapid prototyping by breaking down data processing and featurization into modular components. This modular design allows researchers to reuse core building blocks across different networks and easily add new features. It also simplifies the integration of various datasets, facilitating the training of models like RF3 on a diverse set of biomolecular structures.
4. The framework supports scalable training of biomolecular models. AtomWorks shares most of its code across different networks, allowing researchers to repurpose existing components and improve common operations. This efficiency is demonstrated by the ability to process large batches of data quickly, such as processing a 10,000-token batch through the LigandMPNN pipeline in the time it takes for a single forward/backward pass.
5. AtomWorks is accompanied by industry-grade testing and comprehensive documentation. This ensures that the framework is reliable and easy to use, even for researchers without extensive software development experience. The documentation includes worked examples illustrating how to develop pipelines for various biomolecular modeling tasks.
6. Using AtomWorks, the authors trained RosettaFold-3 (RF3), an all-atom biomolecular structure prediction network. RF3 incorporates novel features such as implicit chirality representations and atom-level geometric conditioning, which improve its performance on tasks like predicting chiral ligands and fixed-backbone conformations.
7. RF3 simplifies dataset integration by supporting direct loading from raw crystallographic information files (CIF). The authors introduced new distillation datasets, including a nucleic acid complex distillation set and an RNA distillation set, to enhance the model's training. Additionally, RF3 includes a disordered distillation set to address issues with hallucinated secondary structures.
8. RF3 accurately adheres to specified stereochemistry out-of-the-box, without requiring inference-time guidance. It represents stereochemistry by the sign of the angles formed by atoms surrounding each chiral center and uses data augmentation techniques to improve chirality handling. As a result, RF3 predicts the correct chirality for 88% of ligand chiral centers in the test set, compared to 84% for AlphaFold3 and 76% for Boltz-2.
9. RF3 enables flexible user control through arbitrary atom-level conditioning. Users can specify distances between atoms to incorporate experimentally derived constraints, perform protein-ligand docking, or fold proteins around specific ligand conformers. This feature significantly improves the accuracy of protein-ligand interface predictions.
10. RF3 narrows the performance gap between existing open-source structure prediction models and AlphaFold3. It demonstrates competitive performance on various tasks, such as predicting protein-protein interfaces, protein-ligand interactions, and mixed L/D peptides. When trained on data up to January 2024, RF3 shows further improvements in performance.
11. The authors also trained ProteinMPNN and LigandMPNN using AtomWorks, demonstrating comparable performance to the original models. This highlights the versatility of the AtomWorks framework in supporting different biomolecular modeling tasks.
12. The AtomWorks framework and RF3 model are released with curated training data, code, and model weights, making them accessible for further research and development in the field of biomolecular modeling.
💻Code: https://t.co/o8iMliCbTx
📜Paper: https://t.co/3SdkXCm8dy
#BiomolecularModeling #AtomWorks #RF3 #StructurePrediction #ProteinDesign #OpenSource #MachineLearning #DeepLearning #ComputationalBiology
Excited to share our preprint “BoltzDesign1: Inverting All-Atom Structure Prediction Model for Generalized Biomolecular Binder Design” — a collaboration with @MartinPacesa, @ZhidianZ , Bruno E. Correia, and @sokrypton.
🧬 Code will be released in a couple weeks
De novo design of miniprotein agonists and antagonists targeting G protein-coupled receptors
🚀 New preprint from David Baker!🚀
1. This paper introduces a computational and experimental approach for designing miniproteins targeting G protein-coupled receptors (GPCRs) with high affinity, potency, and selectivity. The method combines RFdiffusion, MetaGen, and Receptor Diversion (RD) microscopy-based high-throughput screening.
2. Two design strategies are employed: (1) Motif-directed RFdiffusion to target specific receptor hotspots for deep pocket interaction. (2) MetaGen, utilizing diverse structural scaffolds generated from AlphaFold-derived metaproteomes.
3. The Receptor Diversion (RD) screening platform operates directly in human cells, eliminating the need for receptor purification and ensuring compatibility with complex cellular environments. This technique enables efficient screening of up to 100,000 designs.
4. Designed miniprotein agonists successfully activated MRGPRX1, a GPCR associated with itch and pain, with two binders demonstrating full and partial agonistic activity, supported by cryo-EM structures confirming their binding accuracy.
5. The study also developed antagonists targeting CXCR4, GLP1R, GIPR, GCGR, and CGRPR, with nanomolar to picomolar affinities. Notably, the CGRPR antagonist dC2_049 displayed exceptional selectivity and potency.
6. Cryo-EM structures confirmed the designed antagonists’ ability to sterically occlude binding sites and prevent receptor activation, offering new opportunities for GPCR-targeted therapeutics.
7. The successful combination of computational design and high-throughput screening demonstrates a promising route for generating novel protein-based therapeutics targeting GPCRs.
📜Paper: https://t.co/9pKu3X7q0k
#GPCR #ProteinDesign #DrugDiscovery #RFdiffusion #MetaGen #ReceptorDiversion #Bioinformatics #MachineLearning #AI
Announcing Neo-1: the world’s most advanced atomistic foundation model, unifying structure prediction and all-atom de novo generation for the first time - to decode and design the structure of life 🧵(1/10)
@WallStreetMav This is painful to watch. I think no matter which position you have in this, at least we can agree that this should have at least been done behind closed doors.