Assistant Professor in psychiatry, genetics and compmed at UCLA; mom of three; lover of forests and mountains and rain and running; recovering logician
Announcing Misión Origen, a Latin American biobank for severe mental illness that we started building: it will include 50k case and 50k control participants from Colombia!
Here is a video for potential participants we put together, if you’d like to see: https://t.co/9LQXmq3oA5
Excited to see our work in AJHG! Thank you to our amazing co-authors, UCLA Institute for Precision Health, UCLA CTSI, and our collaborators for the support with this work. Above all, we are grateful to the patients and ATLAS biobank participants at @UCLAHealth.
🧬New today!
📄Inclusion bias affects common variant discovery and replication in a health-system linked biobank
🧑🤝🧑 @loldeloo@apimplaskar@bpasaniuc & co
https://t.co/tcU7jJjwzt
🚨 Preprint 🚨 SSRI #antidepressant users report on whether drugs ‘helped’ them.
In @uk_biobank’s 2nd Mental Health Q,
- 20k respondents reported on
- 4 SSRI antidepressants, answering ‘yes’ or ‘no’ to whether specific drugs helped
What do these reports capture?
1/7
1/n
New preprint alert! 🚨🧬✨
The question we asked; is the genetic architecture of a complex trait different at the extreme ends of the distribution (i.e. tails) or similar to that in the main part of the trait distribution?
#ASHG2024#ASHG24 --> check out poster 4051T!
🧵👇🏾
1/n
New preprint alert! 🚨🧬✨
The question we asked; is the genetic architecture of a complex trait different at the extreme ends of the distribution (i.e. tails) or similar to that in the main part of the trait distribution?
#ASHG2024#ASHG24 --> check out poster 4051T!
🧵👇🏾
We are thrilled to announce two open rank faculty searches @UofUGenetics: a broad search for all areas of genetics research (https://t.co/fz3fKZYalB)
& one focused on computational genomics faculty (https://t.co/74HmsEUTna).
This launches a multi-year recruiting initiative!
(1/4)
Can we use genetics to predict which patients with depression will go on to develop bipolar disorder? Are depression and bipolar genetically different?🧬🧠
Today, RGC has published an important paper in @Nature today, describing an analysis of close to a million human exomes (n=983,578) as a single variant call set (!). This is the largest and most diverse rare variant database created so far. This impressive feat is accomplished by a large @RegeneronDNA team led by my wonderful colleague @suganthibala.
@SunKat_y et al. Nature 2024
https://t.co/JiJQxCFKBT
What kind of insights can we learn from sequencing ~980k exomes? Below is a summary of the major findings from the paper.
Background of RGC
Regeneron Genetics Center (RGC) was established in 2014 just on time when major pharma companies started entering into the human genomics playfield. Last year, RGC celebrated its 10th year anniversary. I've written about the origin story of RGC before (https://t.co/LoZRQOpkK1).
The business model of RGC is simple and efficient. It collaborates with academic institutions across the world and provide sequencing as free service in exchange for access to genotypic and phenotypic data.
The first successful collaboration was made with Geisinger Health system (GHS) to sequence 100,000 individuals, which was soon followed by an avalanche of large collaborations. Some of our largest collaborators include UK Biobank (N=500k), GHS (N=175k) and Mexico City Prospective Study (N=150k). Today, RGC has more than 300 collaborations around the world. Just a few months ago, it surpassed the milestone of 2 million exomes. What is described in the current paper is only a fraction of that sample.
Diversity of samples
The 980k exome dataset come from a diverse set of samples. 23% (n=190k) of the participants are of non-European ancestries, the largest proportion to date for any similar datasets created so far. This includes both outbred populations and special populations enriched with communities with long-standing cultural history consanguineous and endogamous unions.
When it comes to human genetics, diversity is the key to making discoveries. Almost everyone agrees, and the field is embracing it now. But RGC is way ahead of the game. Just a few months ago, RGC partnered with other companies and laid the first foundational stone of what will become in a few years from now the world's largest genomics resource comprising half a million African Americans and Africans (https://t.co/JE0Yycm8E1).
Variant survey
Human genome is ~3 billion base pairs long. ~1% of which (~30 million base pairs), containing exons, is targeted by exome sequencing. By sequencing 980k exomes, the authors have captured ~16.5 million unique variants. That is, on average, one per every two base pairs across the exome.
The main goal of concentrating on exomes is to capture deleterious spelling errors in the genome, resulting in either loss or substantial decrease or, sometimes, increase in gene function. The authors have identified
- ~1.1 million predicted loss of function variants (pLOFs), ~50% of which are singletons (that is, seen in just one individual)
- ~10 million missense variants, 40% of which are singletons.
As expected, African ancestry groups had more variants (18% more) than any other ancestry group.
Footprints of selection
pLOFs in the human genomes are like bullet holes in aircraft returning from war. The genes untouched or rarely hit by the pLOFs are the most critical genes, without which life is probably impossible.
Studying ~980k exomes, the authors have identified ~4000 genes that are depleted of pLOFs, suggesting they are indispensable. For more than 20% of these genes, we are learning their critical requirement for normal life for the first time. Previous datasets were not able to quantify their mutation constraints because of the shorter length. Most of these genes were not linked to a human disease yet. The current list will inspire many Mendelian discoveries in the near future.
Regional selection
We have 10 times more missense variants than pLOFs, which means we can zoom into within genes and study which parts of a gene are indispensable and which parts aren't.
Not all parts of a protein are critical, but some parts are. For example, DNA binding regions of transcription factor protein, catalytic sites of an enzyme protein, transmembrane domains that forms the pore of channel proteins etc. With a knowledge of ~10 million missense variants from 980,000 humans, such critical regions are now starting to light up, illuminating the most crucial regions of proteins. For example, here is a trace of missense tolerance across different domains of cancer gene KRAS. Human genetics shows that the first 80 amino acids as the most critical region of KRAS, falling under the top 1 percentile of regional missense constrain metric.
Human knockouts
The function of a gene in an organism is understood, typically, by studying the phenotypic consequences of deleting the gene. We cannot do such experiments in humans. But fortunately, Nature has already done this mutagenesis experiments for us. By studying naturally occurring human knockouts, we can assess the consequences of completely inhibiting a gene. This is crucial data for drug developers, as it informs about safety of drugs that act by inhibiting a gene or its product.
Studying the pLOFs across 980k humans, the authors have found 4.686 genes with at least one human knockout, suggesting that a life without these genes is likely possible. In line with that, the authors find that these genes are the ones that were mutationally least constrained (that is, they are enriched for pLOFs). For >1700 genes, we are learning for the first time humans completely lacking these genes do exist in this world. This is an incredible resource for drug development.
Clinical genetics insights
One of the most important use case of reference variant databases is to help clinical geneticists to identify disease causing variants in the patients. Historically, variant databases have been biased towards European populations. As a result, clinical geneticists struggle when they study exomes of non-European ancestry patients and often label the suspected variants as variants of unknown significance (VUS), because of a lack of proper reference database.
Cross-referencing the clinvar database with RGC dataset, the authors find European ancestry groups had more variants labelled "pathogenic" in Clinvar than African ancestry groups. Conversely, African ancestry groups had more VUS than European ancestry groups. This is not because Africans are protected from pathogenic variants, but simply reflect current databases are ignorant to clinically important variants in non-European ancestry individuals. With growing diverse databases such as the current one from RGC, the situation will soon change.
Conclusion
RGC has created one of the largest reference database for studying human exomes. The implications of this resource are many, spanning all areas of human biology from basic science to drug discovery.
Congrats to all my colleagues (@SunKat_y et al.) on this incredible accomplishment. And thanks to all RGC collaborators and research participants without whom such a dataset wouldn't exist.
[https://t.co/HewhGoAiKF] Cancer biomarker discovery is historically mutation-centric, often neglecting the key role of genomics instability. Recently, we asked ourselves how do mutations and copy number alterations (CNAs) determine prognosis and tropism? 😋😋1/n
I've written the first part of a chapter on the heritability of IQ scores. Focusing on what IQ is attempting to measure. I highlight multiple paradoxical findings demonstrating IQ is not just "one innate thing".
https://t.co/MVJSmWnTTL
I'll summarize the key points here. 🧵
The UCLA Goodman-Luskin Endowed Postdoctoral Fellowship in Microbiome Research ($100K in salary + supplies) deadline is June 1. Get in touch if you would like to write a proposal together!
And, get in touch even if your timeline is different - I am looking for a postdoc!
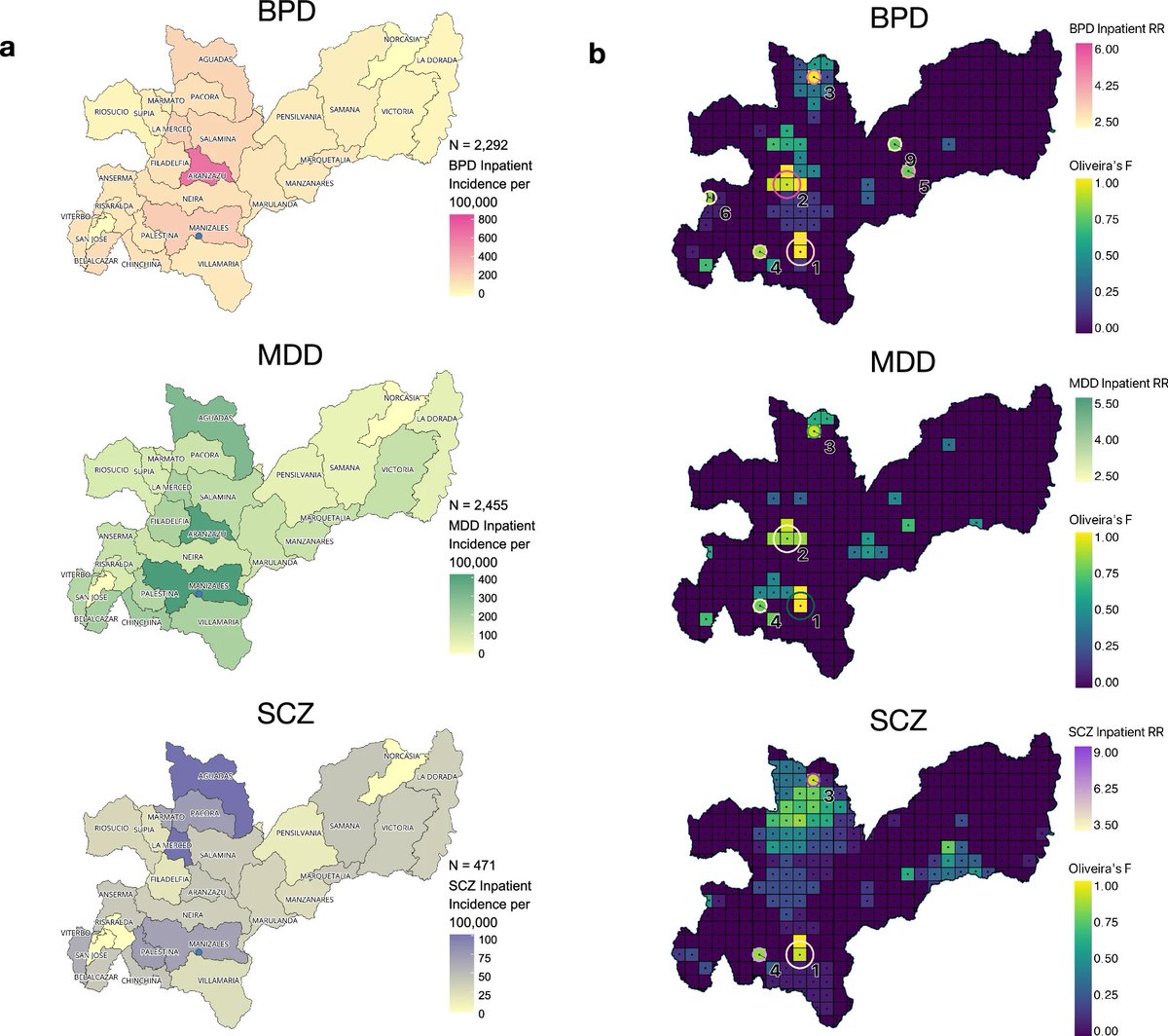
#Geospatial Investigations in Colombia reveal variations in the distribution of mood and psychotic disorders by Song, @loldeloo et al in @CommsMedicine
Inequitable geographic accessibility of specialty mental healthcare services.
Targeting resources to identify severely ill individuals living in the observed hotspots could further address treatment inequities and enable investigations to determine the factors generating these hotspots
https://t.co/pV8xVXWZej
See more articles in the collection: "Geospatial analysis for improved understanding of health inequalities." https://t.co/NyNjXXgm2s
This has been an amazing team effort of many - including collaborators from the Universidad de Antioquia, Clínica San Juan de Dios in Manizales, UCLA and more
Two takeaways:
1) We observed inequities in access to healthcare: many individuals requiring only outpatient treatment may live too far from the clinic to access healthcare
2) For inpatients, we found several hotspots in the region (one with among the highest frequencies of bipolar disorder reported worldwide!). These are candidates for further research to identify genetic or environmental risk factors
Using geospatial investigations of electronic health records from Colombia we found variations in the distribution of mood and psychotic disorders
Check it out! 👇
https://t.co/2iDcEIkCak