When I created Twitter account, I promised myself not to post anything unrelated to science. However, the current situation goes beyond all conceivable limits. Unfortunately, I can't change anything, but at least I can say it here: THE WAR MUST BE STOPPED.
@bravo_abad And in addition to a simple PCA baseline, one may use a platora of already pretrained models (i.e. genome FM - https://t.co/A2Pkch1YEc, RNA-seq FM - https://t.co/ID7cdNRkC6, etc.)
It's amazing how simple it is to use AI for genomic variant interpretation nowadays!
Example: fully-agentic study of deletion in BMPR1A promoter
https://t.co/j9D6tdbOVg
What's missing to make it routine for clinics & pharma? More accurate and reliable models.
Just converted hemoglobin promoter into something active* in hepatocytes (HepG2) with Codex and GI skills from ClawBio!
Full post: https://t.co/yXJDPxwxlx
*active == predicted to be active by our expression model. It is SOTA in expression prediction, but we never know for sure.
Thank you, @acarroll_ATG , for the thoughtful write-up on genome foundation models. I particularly appreciated the distinction between a large pretrained model and a representation that is actually useful for biology.
I agree with the premise that in genomics, scale is not sufficient. What matters is what the model learns, from which sequences, under which objective, and against which biological constraints.
I see the current state as an intermediate stage.
Today, DNA foundation models can be useful as priors. They can improve initialization, sample efficiency, and adaptation when labels are scarce. But they do not automatically replace strong supervised sequence-to-function models on well-defined regulatory tasks.
The next step is not simply larger embeddings.
A more useful direction is a long-context, DNA-only pretrained backbone that learns a broad sequence prior such as coding constraints, repeats, evolutionary structure, gene architecture, motif structure, regulatory syntax, and long-range dependencies. But that backbone should not remain a frozen embedding generator. It should be post-trained jointly with measured biology.
In such a system, the DNA backbone remains trainable and is combined end-to-end with additional encoders for chromatin accessibility, TF binding, histone marks, RNA-seq, splicing, 3D contacts, perturbation readouts, single-cell state, phenotype, and experimental feedback from designed sequences.
Different decoders can then specialize the same genomic prior for different biological functions, including expression, variant effects, enhancer-promoter logic, splicing, regulatory sequence design, cell-type-specific response, disease risk, and therapeutic construct optimization.
This is where several important lines of work seem to converge.
David R. Kelley and colleagues on Enformer and Borzoi for sequence-to-function modeling trained on rich functional genomic readouts with sufficient regulatory context.
DNA-only encoder and generative pretraining across genomic diversity, with long context and single-nucleotide resolution.
@anshulkundaje and colleagues' models to recover regulatory grammar, motif syntax, and causal sequence logic.
The next useful systems should combine these ideas: DNA-scale pretraining, joint post-training on functional genomics, modality-specific encoders, function-specific decoders, and evaluation by biological utility. That means variant interpretation, sequence design, cross-cell-type generalization, perturbation prediction, uncertainty, interpretability, and experimental validation.
We are also starting to make some of the infrastructure around genomic language models and DNA language models easier to use and adapt, intending to make it accessible to a wider research community: you can try here https://t.co/XAdgpDslIP
Hopefully, this will help clarify which assays, modalities, fine-tuning recipes, and biological questions actually benefit from this approach.
The service also supports gene finding / predicted transcript intervals from DNA sequence. Have an unannotated genome? Try to use it. More about it in our papers: https://t.co/44DVJNHeni
4/n
New browser tool for functional DNA annotation: seq2expression (like aGenome, but more flexible), ab initio gene annotation,splice sites, promoters, etc!
Try it: https://t.co/bAD6ilmvws
Looking for feedback (@ensembl, @scverse_team, @GeneticsSociety, @genomicsengland)!
1/n
The idea is to make it easy to explore questions like:
• How does predicted expression change across cell types or conditions?
• How might sequence changes affect predicted expression?
• Can model-based annotations help prioritize regions for follow-up work?
3/n
What if you could ask an AI model what a DNA sequence does?
We built a demo for exactly this.
Try it here: https://t.co/0GCCch7wMO
You provide a DNA sequence + a plain-English biological context, like a cell type or assay condition.
The model predicts gene expression in that context.
It also supports promoter prediction, enhancer prediction, splice-site prediction, gene finding, and sequence annotation.
This is still early, but the direction is clear:
DNA models will make biology more queryable, predictive, and eventually programmable.
Would love feedback from people in genomics, gene regulation, synthetic biology, variant interpretation, and AI.
#AI #Genomics #Biotech #SyntheticBiology
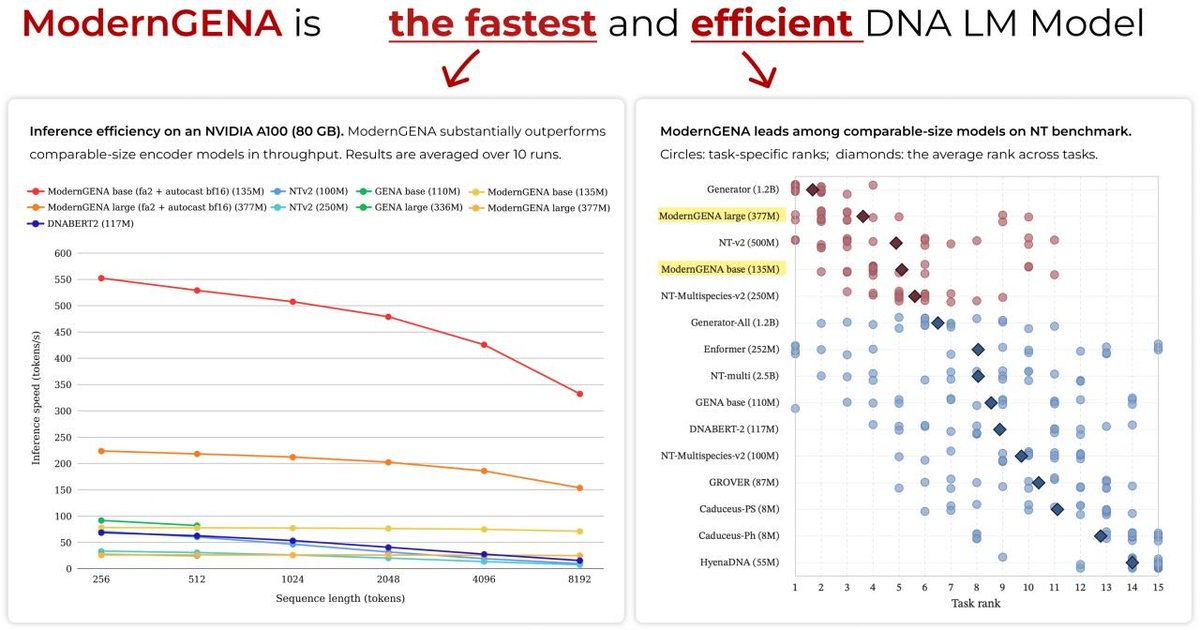
We will present our new DNA Language model, ModernGENA (https://t.co/m3bZFRrpRN), at #ICLR workshops on Monday, April 27!
Our posters will be presented at #ICLR workshops on Monday, April 27:
FM4Science 4:00 PM
MLGenX 3:40 PM
Gen2 4:10 PM
We welcome discussions on DNA LMs!
In our paper (https://t.co/RzeVmVQnCD) we study how modern Transformer improvements transfer to genomics. We show that the ModernBERT-based architecture behind ModernGENA achieves the high throughput while maintaining competitive performance.
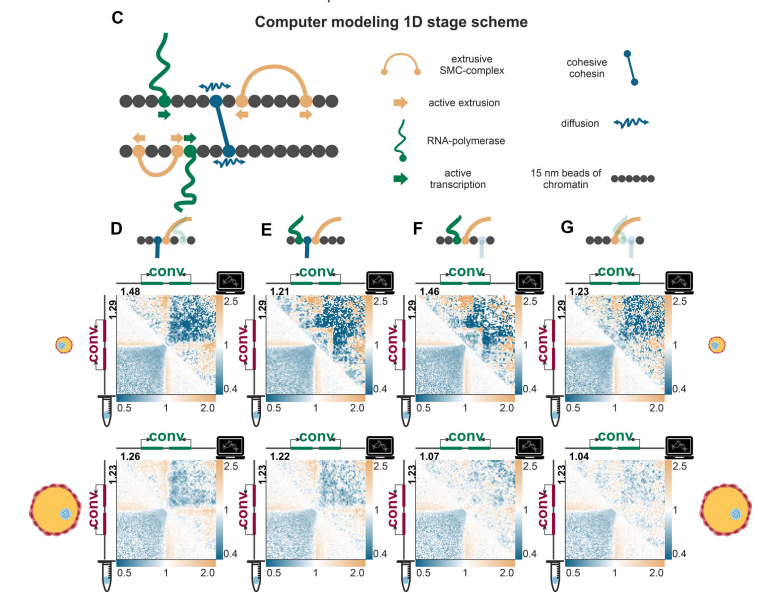
I think it aligns well with what @dekker_lab@golobor , and others showed before for other cell types - but in LBC, hypertranscription results in an extreme, amplified version of these general principles
1/
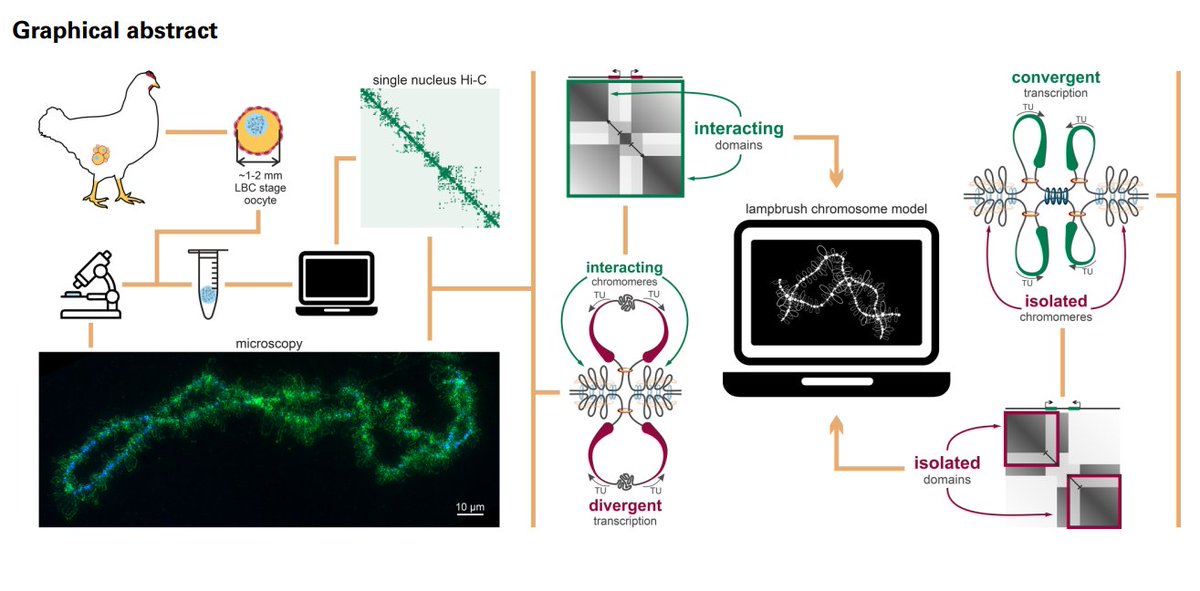
Our paper just came out in Nucleic Acids Research! 🎉
We study one of the most fascinating chromatin systems — lampbrush chromosomes (LBCs) in chicken oocytes.
🔗 https://t.co/vZxury709S
8/
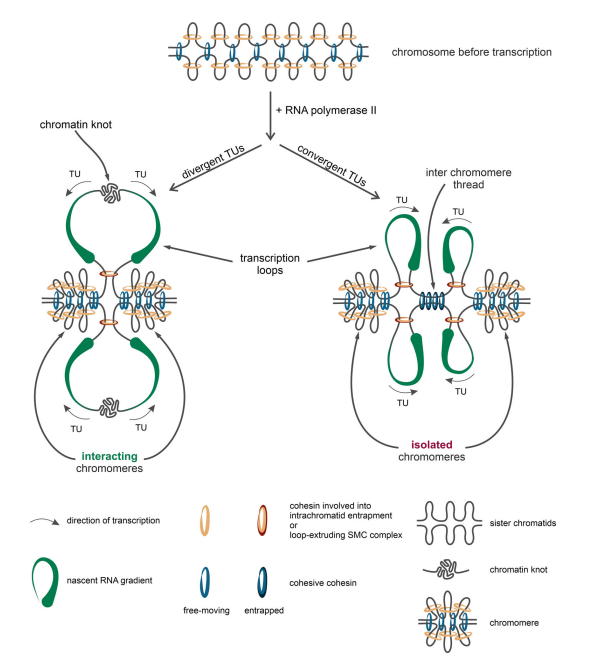
A key insight:
👉 Gene orientation matters
Convergent genes (→ ←):
→ trap cohesin between them
→ form strong insulation boundaries
This explains domain formation without CTCF
10/
Huge thanks to collaborators from Saint-Peterburg University (Alla Krasikova’s group), experimental (Maria Gridina and co) & bioinf/biophys (Timofey Lagunov and co) teams! This was a great pleasure working with them!