🚨Publication alert🚨 My first-author PhD paper is out today in Nature Genetics https://t.co/pq2Nocftsx
Tumors aren’t one entity — they’re made of coexisting cell states with distinct vulnerabilities. We show that targeting complementary states simultaneously in DMG is key.
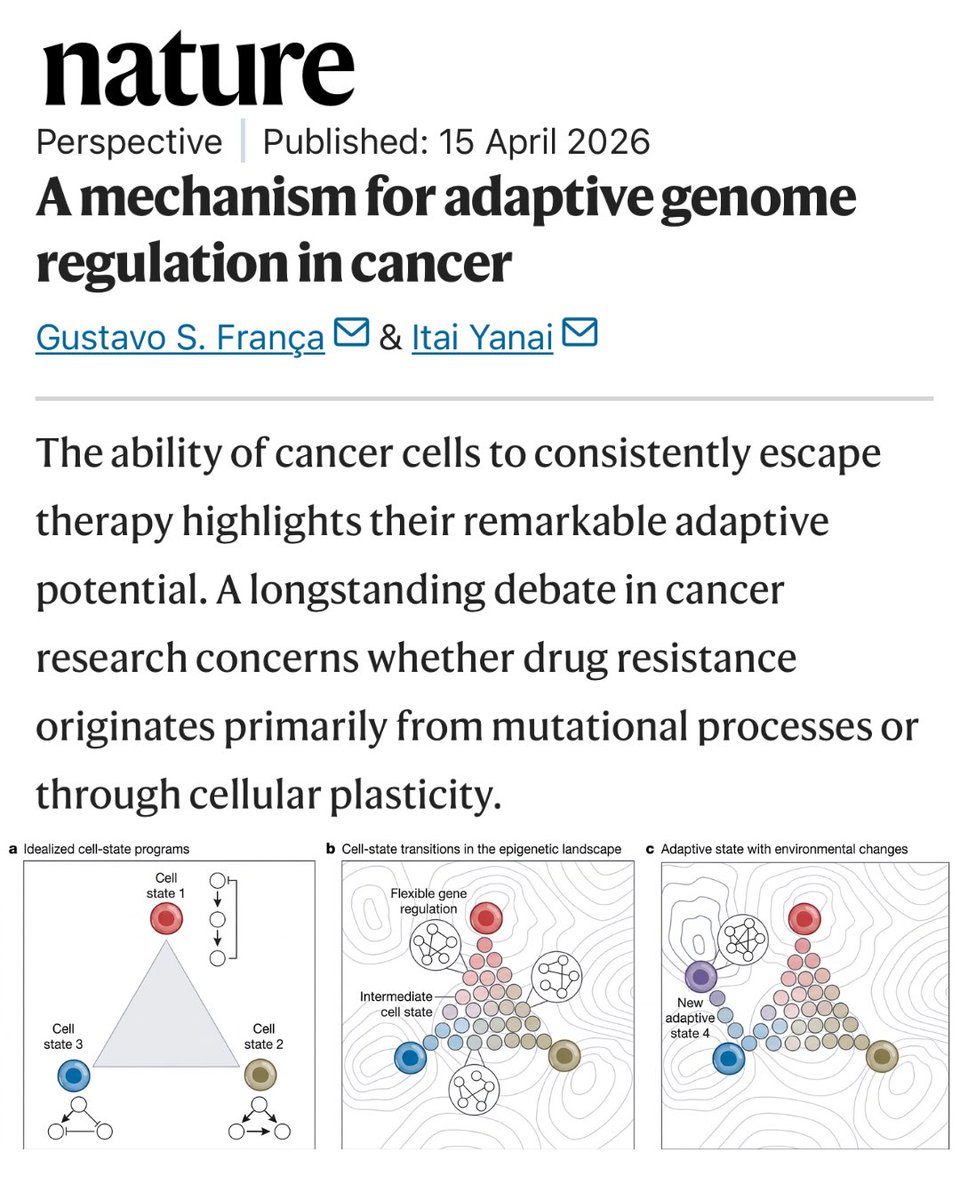
New research in Nature suggests cancer cells can learn to resist therapy, not by mutating, but by reprogramming themselves.
In #lungcancer, resistance to targeted treatments is a significant challenge. Understanding how this happens is an important step in the search for better treatment
🔗 https://t.co/RvSA6FljUj #LCSM
How does a cell learn? In our new perspective in @Nature we propose a model where the properties of the AP-1 family of transcription factors – stress-induced feedback, regulatory combinatorics & cellular memory – encode a mechanism for cellular learning. https://t.co/Hzmq319EaD
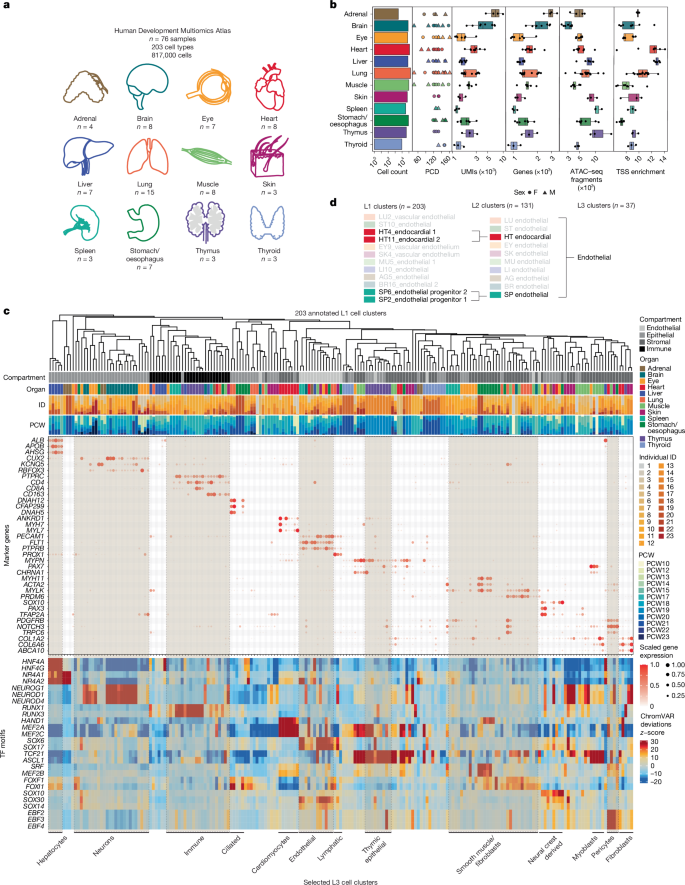
Motif syntax governs cell-type-specific chromatin accessibility and provides a foundational resource for decoding cis-regulatory logic and interpreting genetic variation during human development @Nature@anshulkundaje@WJGreenleaf@Stanford
https://t.co/kTQIhM3ImP
Excited to share our latest work:
https://t.co/8x8pQFpFeI
Huge effort led by @AminNoorani@UHN_Research@mcgillu@broadinstitute
We generated a single-cell chromatin accessibility atlas across multiple tissues, highlighting how epigenomic landscapes can trace cell-of-origin.
Excited to share my postdoc work with @JD_Buenrostro now out in @Nature!
"Epigenetic memory of colitis promotes tumour growth"
https://t.co/6703aeQlLG
We wanted to understand how transient inflammation can create a long-lived increase in cancer risk, even after full recovery 🧵
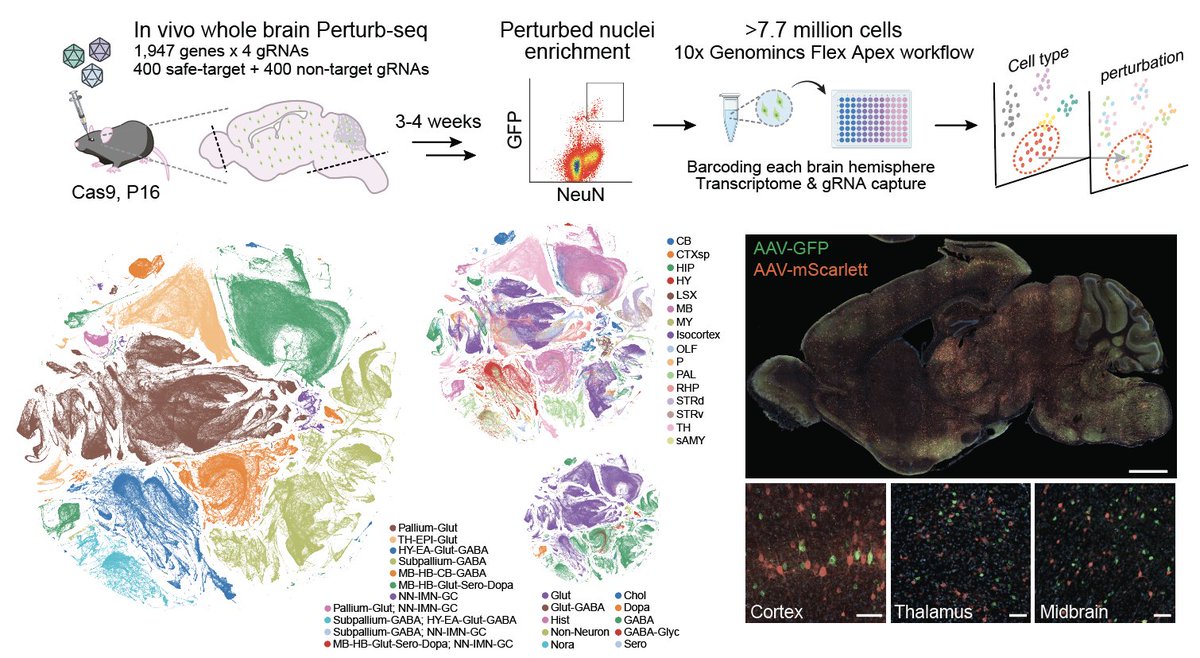
📢 Preprint: we present a whole-mouse-brain in vivo Perturb-seq atlas, 7.7 million cells, 1947 disease-associated perturbations, moving toward direct readout of how human genetics rewires cell states & circuits in vivo. Grateful for the Team! @NVIDIAHealth https://t.co/01c1KFuLFw
I am really excited to share that our review on preclinical models of breast cancer metastasis is out in npj Breast Cancer! We provide a comprehensive overview of the major preclinical models used to study breast cancer metastasis @sflomos@NadineDarwiche https://t.co/bOTfLfzxBK
What is sci-Plex-GxE? A highly scalable platform that pairs pooled CRISPRi/a screens with combinatorial indexing scRNA-seq to measure genotype × environment (GxE) effects across conditions (here, graded T cell pressure, ~520 kinase perturbations × 4 E:T ratios × 2 modalities)👇
VIPerturb-seq is based on a simple idea: we associate CRISPR gRNA with a split barcode detected by the combination of a left-hand + right-hand probe
This lets us easily read out very large (genome-wide) gRNA libraries w/probe-based scRNA-seq, like the Flex kit from @10xGenomics
How does histone code work in human brain? Excited to share our new preprint on human brain single-cell histone modification landscape, as part of the BICAN effort in collaboration with Salk/UCI/WUSTL 👉 https://t.co/NRaLaFRLbI
Full dataset will be released shortly. Stay tuned.
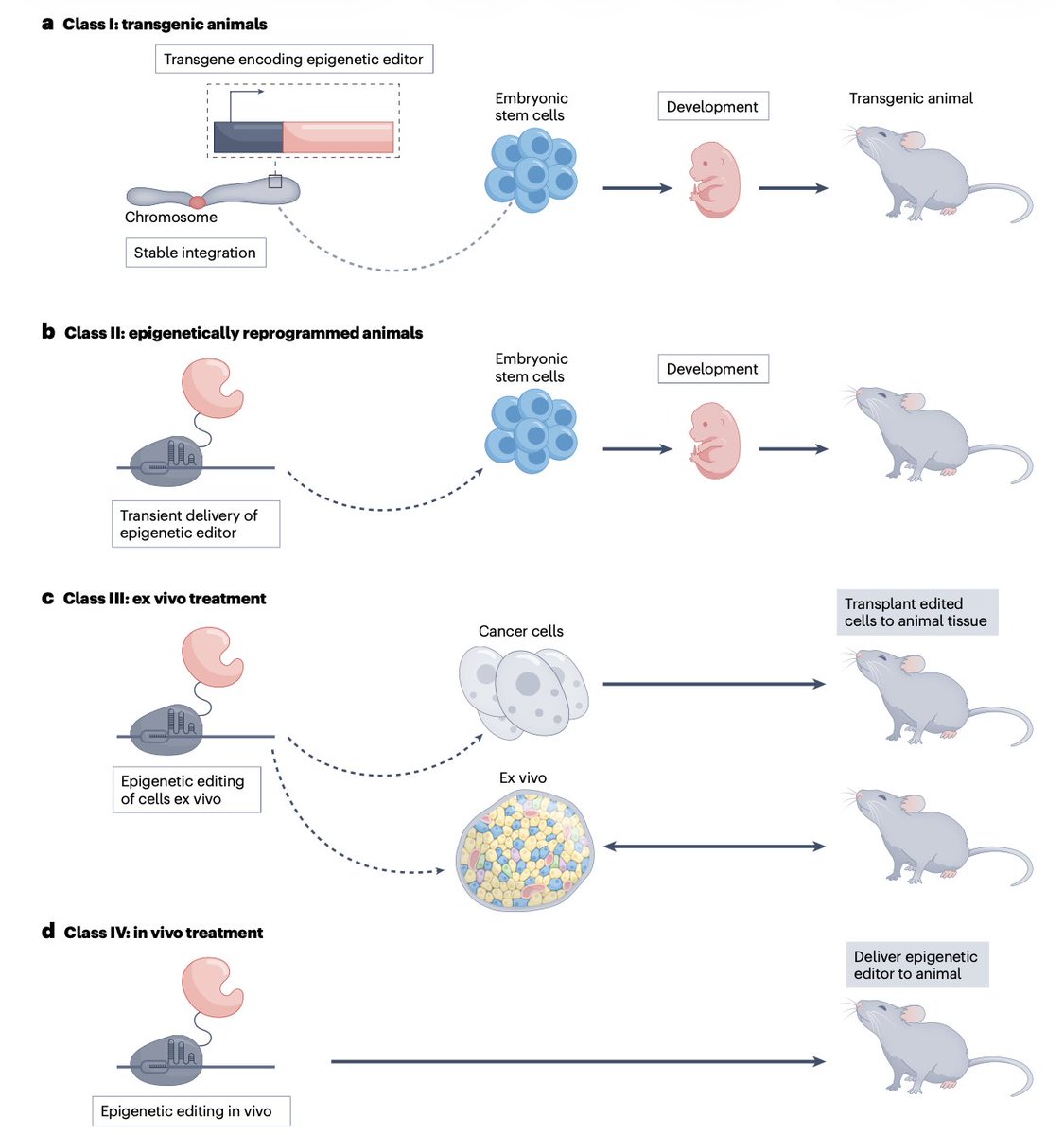
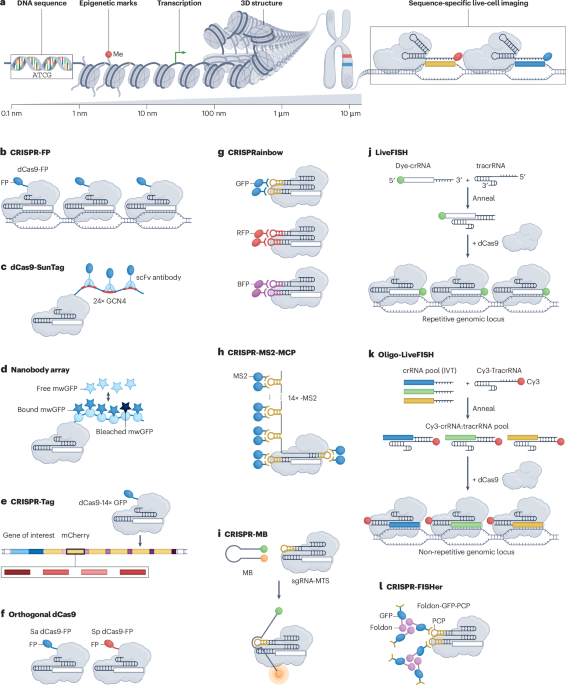
🧬 Epigenetic editing: from concept to clinic.
Epigenetic editing enables durable gene expression reprogramming without altering DNA sequence.
Early concerns on efficacy and specificity are being addressed through improved understanding of epigenetic maintenance, targeting precision, and delivery systems. First clinical trials are now underway.
📖 @NatRevDrugDisc
DOI 👉🏻 10.1038/s41573-025-01323-0
#CánCare #oncology #cancer #epigenetics #precisiononcology
AlphaGenome: Decoding the dark matter of the genome with a unified deep learning model
More than 98% of human genetic variation lies outside protein-coding regions. These "non-coding" variants can disrupt gene regulation in remarkably diverse ways: altering chromatin accessibility, shifting 3D genome architecture, modifying splicing, or changing expression levels—often in tissue-specific patterns. Yet existing computational models face a fundamental trade-off: either they capture long-range regulatory interactions (like distant enhancers) but blur fine-scale features, or they achieve nucleotide resolution but miss distal context. And most specialize in a single modality, leaving users to stitch together predictions from many separate tools.
Žiga Avsec and coauthors at Google DeepMind present AlphaGenome, a model that sidesteps these trade-offs. It takes 1 megabase of DNA as input and predicts ~6,000 genome tracks—spanning gene expression, splicing (sites, usage, and junctions), chromatin accessibility, histone modifications, transcription factor binding, and 3D contact maps—at up to single-base-pair resolution.
The architecture combines a U-Net backbone with transformer blocks: convolutions capture local motifs essential for splice sites and TF footprints, while transformers model long-range dependencies like enhancer–promoter interactions. Training uses a two-stage approach—pretraining on experimental data followed by distillation from an ensemble of teachers using mutationally perturbed sequences—yielding a single model that scores variants across all modalities in one pass.
The results are striking: AlphaGenome achieves state-of-the-art performance on 25 of 26 variant effect prediction benchmarks, including a 25% improvement in predicting eQTL direction over the previous best model. It outperforms specialized models on their own tasks—beating SpliceAI-class methods on 6 of 7 splicing benchmarks and ChromBPNet on accessibility QTLs. Critically, the multimodal outputs enable mechanistic interpretation: for oncogenic mutations near the TAL1 gene in T-cell leukemia, AlphaGenome simultaneously predicts neo-enhancer formation (increased H3K27ac), chromatin opening, and elevated gene expression—recapitulating experimentally validated mechanisms.
This points toward a future where interpreting non-coding variation no longer requires assembling a patchwork of specialized models. A unified framework that jointly predicts molecular consequences across modalities could accelerate rare disease diagnostics, guide therapeutic oligonucleotide design, and help prioritize variants in GWAS loci—moving us closer to truly reading the regulatory code written in DNA.
Paper: https://t.co/3WzrnGNUSw
🧬 Another super exciting new work from the lab: Decoding the gene regulatory landscape through multimodal learning of protein–DNA interactions.
👉 Link to preprint: https://t.co/SLWIYeLnUG
In this work, we introduce Chromnitron, a biologically grounded multimodal foundation model that decodes the global gene regulatory landscape by hundreds to thousands of proteins binding in the genome.
⁉️ Why is this protein-centered view of regulatory genomics so important? DNA sequence 🧬provides the same genetic blueprint, but the regulatory logic that gives rise to diverse cell types and states – in both health and disease conditions – is executed by the 100s-1000s of chromatin proteins that differentially read the DNA sequences to establish cell-type-specific gene expression programs.
🤖 Chromnitron is specifically developed to decode this cell-type-specific regulatory logic. Designed to learn from the first principles that determines protein–DNA interactions in the cell, Chromnitron integrates 1) genomic DNA sequence, 2) cell-type-specific chromatin states, and importantly, 3) protein sequence-encoded structural features.
👨🏻💻 Through large-scale pre-training & fine-tuning with a finely curated atlas of high-quality datasets, Chromnitron learns the shared and protein-specific mechanisms of protein–DNA interactions, and thus can accurately predict the binding landscapes across hundreds of proteins in unseen cell types.
💡Chromnitron isn’t just accurate and cell-type-specific prediction — it is a discovery engine! With Chromnitron:
we identified novel regulators of T cell exhaustion (ZNF865 & ZNF766);
we reconstructed the dynamic regulatory landscapes of how global chromatin proteins orchestrate neurodevelopment.
🔮 Chromnitron represents a significant step toward predictive, interpretable, and mechanistic understanding of gene regulation and cell fate determination. We expect Chromnitron to accelerate discovery and engineering in regulatory genomics, particularly in human biology, and empower therapeutic opportunities.
🙏🏻 Last, this is another wonderful collaboration with Dr. Aristotelis Tsirigos @artsinyc, and led by the brilliant Drs. Jimin Tan @tan_jimin, Xi Fu @alexanderfuxi, and Xinyu Ling @Dennislxy, and many more co-authors and friends who helped make this work truly possible!
👉 Find out more about the model and discoveries (e.g. architecture, interpretability, benchmarking, generalizability, etc): https://t.co/SLWIYeLnUG
#MultimodalAI #GeneRegulation #TranscriptionFactor #RegulatoryGenomics #CellFate #AIinBiology