Wow, I think this is the start of mechanism-first protein design.
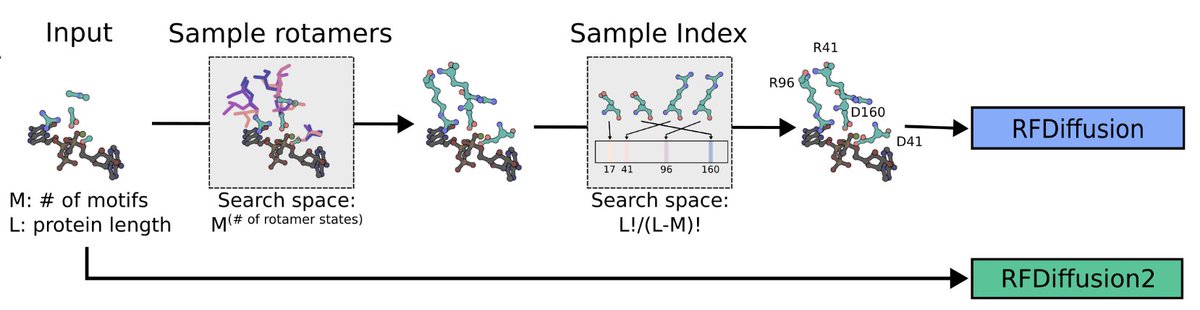
RFdiffusion2 redesigns enzyme creation from the atom up—directly scaffolding active sites from transition state geometries, no rotamers or residue indices needed.
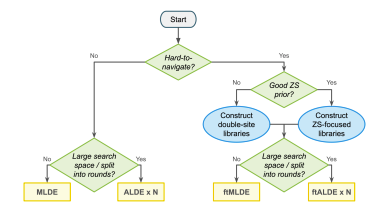
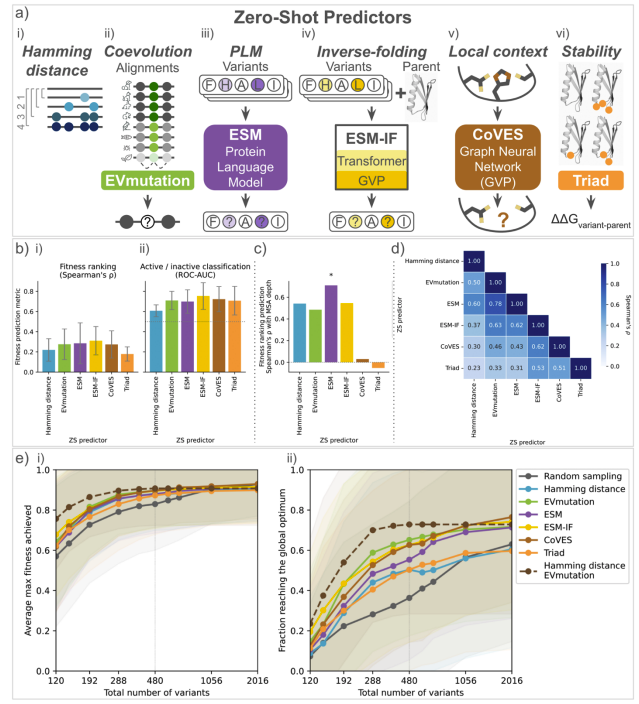
Self-supervised machine learning methods trained on natural sequences and structures are good at separating functional and nonfunctional variants but do not identify the best variants.
We compared the calibration of various machine learning uncertainty estimation methods for protein engineering.
No method excels across all scenarios, and uncertainty-based strategies for optimization often did not outperform methods without uncertainty.
Computational Design of Metallohydrolases
🚀 New preprint from David Baker!🚀
1. This study introduces RFam, a generative AI-based flow-matching model, which enables atomic-level scaffolding of enzyme active sites without predefined sequence positions or rotamer states, overcoming key limitations of previous methods like RFdiffusion.
2. Using density functional theory (DFT) descriptions of active site geometry, RFam designed 96 zinc metallohydrolases, achieving unprecedented catalytic efficiency, with the top design (A1) displaying a kcat/KM of 23,000 M⁻¹ s⁻¹—orders of magnitude higher than prior de novo designs.
3. The A1 enzyme features a novel fold with a highly preorganized active site and an enclosed pocket optimized for substrate positioning and catalytic efficiency, verified using the deep learning tool ChemNet.
4. RFam uniquely scaffolds functional groups rather than backbone motifs, significantly enhancing sampling of sequence space and enabling the generation of diverse and functional protein scaffolds.
5. Experimental validation showed 86 out of 96 RFam designs were soluble, and five demonstrated significant activity. A1 stood out with over 1,000 turnovers and remarkable stability during prolonged catalysis.
6. The study pioneers the integration of quantum chemistry, AI-driven design, and deep learning-based ranking to produce highly active, function-specific enzymes without the need for extensive directed evolution.
7. This approach opens doors to applications in environmental remediation and synthetic biology, as metallohydrolases can be tailored to degrade complex human-made pollutants efficiently.
@samansalike31@ikalvet@woodyahern
📜Paper: https://t.co/hsjyWKiEYB
#EnzymeDesign #Metallohydrolases #ArtificialIntelligence #QuantumChemistry #SyntheticBiology #ProteinEngineering
My team at Deepmind (protein design) is hiring an experimentalist with enzyme expertise. Please RT and/or apply! I'm happy to answer any questions as well. https://t.co/2acSQJX4At
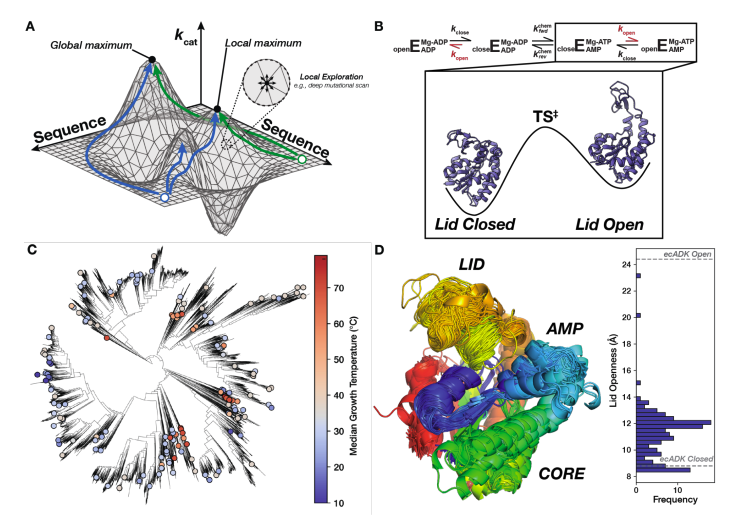
Experimental kcat and KM measurements for hundreds of naturally variants of adenylate kinase.
- thermophilic enzymes are not slower than mesophilic!
- general kcat/KM predictors are bad and easily beaten by models trained on this specific dataset!
Machine learning-guided directed evolution strategies exceeded or at least matched DE performance with the advantages becoming more pronounced as landscapes had fewer active variants and more local optima.
@francescazfl@yisongyue@jsunn_y@kadinaj@francesarnold
Fantastic to see the chemistry Nobel be awarded for this essential work - especially when we use it in our group on a daily basis!
#chemnobel#NobelPrizeChemistry
Computational Stabilization of a Non-heme Iron Enzyme Enables Efficient Evolution of New Function
🚀 New paper from David Baker and Jesse Zalatan!🚀
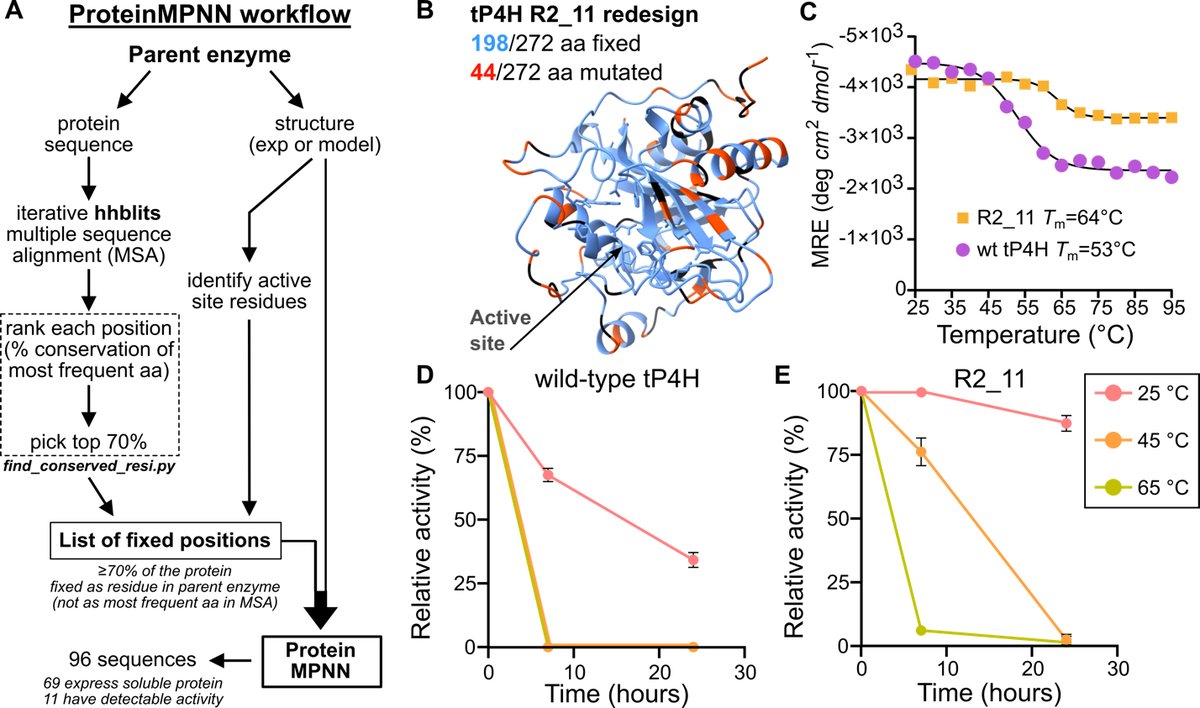
1/ This study demonstrates the use of the deep learning-based tool, ProteinMPNN, to computationally redesign Fe(II)/αKG superfamily enzymes, improving their stability, solubility, and expression while retaining catalytic function.
2/ The redesigned enzymes were successfully evolved for a non-native, remote C(sp3)-H hydroxylation reaction, achieving an 80-fold increase in activity over three rounds of directed evolution compared to just a 6-fold improvement from the wild-type enzyme.
3/ A critical innovation of the study is the use of ProteinMPNN to introduce stabilizing mutations, allowing for more efficient evolution trajectories, saving both time and resources in the process of engineering biocatalysts.
4/ This work showcases the effectiveness of stabilizing enzymes before mutagenesis. The starting stabilized variant provides access to mutations that otherwise would not fold correctly in a wild-type enzyme, enabling new opportunities for enzyme engineering.
5/ The generalizability of the approach is highlighted by successful applications to multiple enzymes within the Fe(II)/αKG superfamily, indicating that ProteinMPNN can be a routine tool in directed evolution workflows for a variety of biocatalysts.
@KieraSumida@UWproteindesign
📜Paper: https://t.co/0EY2M6lkLB
Improved protein binder design using beta-pairing targeted RFdiffusion
🚀 New preprint from David Baker!🚀
• A breakthrough in protein binder design: this study uses RFdiffusion conditioned to generate binders that form precise beta-strand pairings with polar protein targets, yielding binders with superior affinity and specificity.
• The approach focuses on polar regions of target proteins, especially edge beta-strands, addressing the challenge of complementing hydrogen bond donors and acceptors that are otherwise exposed to water.
• Designed binders achieved affinities ranging from 76 pM to mid-nanomolar, targeting therapeutically relevant proteins like KIT, PDGFRɑ, and ALK-2, with no significant off-target binding.
• A co-crystal structure of a binder in complex with the KIT receptor confirms the computational accuracy of this new design method, showcasing near-perfect matching of the design model and real-world interactions.
• This beta-strand focused method not only enhances the range of RFdiffusion’s capabilities but also expands the potential for developing new therapeutic binders targeting polar protein surfaces, such as those in cancer-related pathways.
• The new design approach surpasses traditional methods, particularly in targeting polar regions and edge beta-strands in proteins like ALK-2, ALK-3, and PDGFRɑ, where previous techniques failed to generate effective binders.
• The study demonstrates that beta-strand pairing conditioned designs have higher thermal stability and are more specific in binding compared to earlier models, making them valuable for therapeutic applications like receptor targeting and drug delivery.
@UWproteindesign@SavvidesLab@StevenMBanik@TimothyPJenkins@Green_Ahn@MelBenard86@kf_verstraete@SusanaVazTor@_JosephWatson@martin_toul
📜Paper: https://t.co/nadc5mZzDq
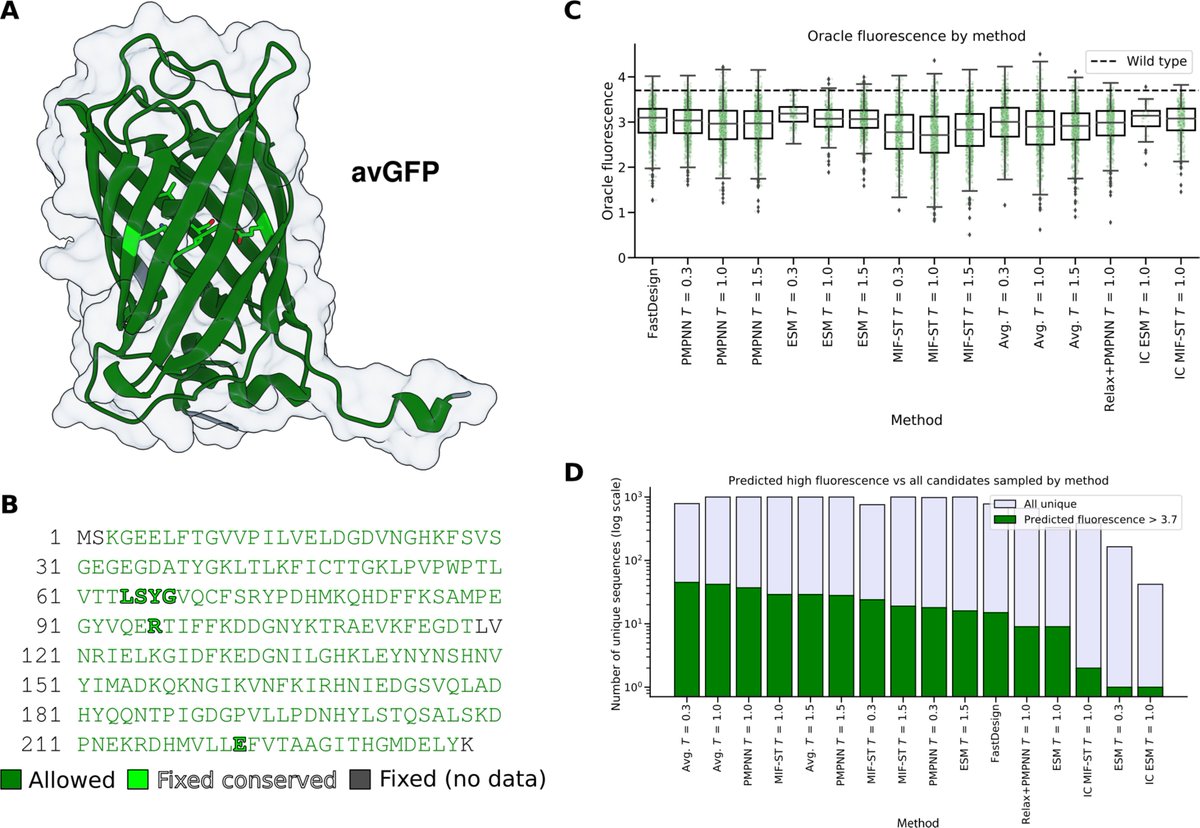
Protein Language Models: Is Scaling Necessary?
- This paper challenges the prevailing belief that scaling up protein language models (pLMs) is essential for better performance, proposing that careful data curation can achieve comparable results at a fraction of the cost.
- The authors introduce AMPLIFY, a protein language model that outperforms state-of-the-art models like ESM2 15B, while being 43 times smaller in terms of parameters and 17 times more efficient in training.
- AMPLIFY’s success is attributed to using high-quality, curated datasets rather than simply increasing model size. This allows for better generalization and less overfitting, particularly in tasks like sequence recovery and protein design.
- By focusing on natural sequence space and eliminating noise from datasets, AMPLIFY reduces computational costs and energy consumption, democratizing pLM development for smaller research labs.
- The paper emphasizes that data quality is more important than model size, with findings showing that models trained on well-curated datasets significantly outperform models trained on larger but noisier datasets.
- AMPLIFY exhibits emergent behaviors in tasks like distinguishing real proteins from non-proteins, even in zero-shot settings. It can also handle intrinsically disordered proteins better than structure-based models like AlphaFold2.
- The authors call for a shift away from scaling as the main driver of improvement in pLMs, advocating for better dataset curation and efficient architectures to build robust, high-performing models.
@apsarathchandar@bnschlz
💻Code: https://t.co/QPdgUBcswf
📜Paper: https://t.co/qFdubhLQ1t
Have you ever wanted to design protein binders with ease? Today we present 𝑩𝒊𝒏𝒅𝑪𝒓𝒂𝒇𝒕, a user-friendly and open-source pipeline that allows to anyone to create protein binders de novo with high experimental success rates. @befcorreia@sokrypton
https://t.co/IPhMFpRgHh
A Computationally Designed Panel of Diverse and Selective Peroxygenases for Terpene Oxyfunctionalization
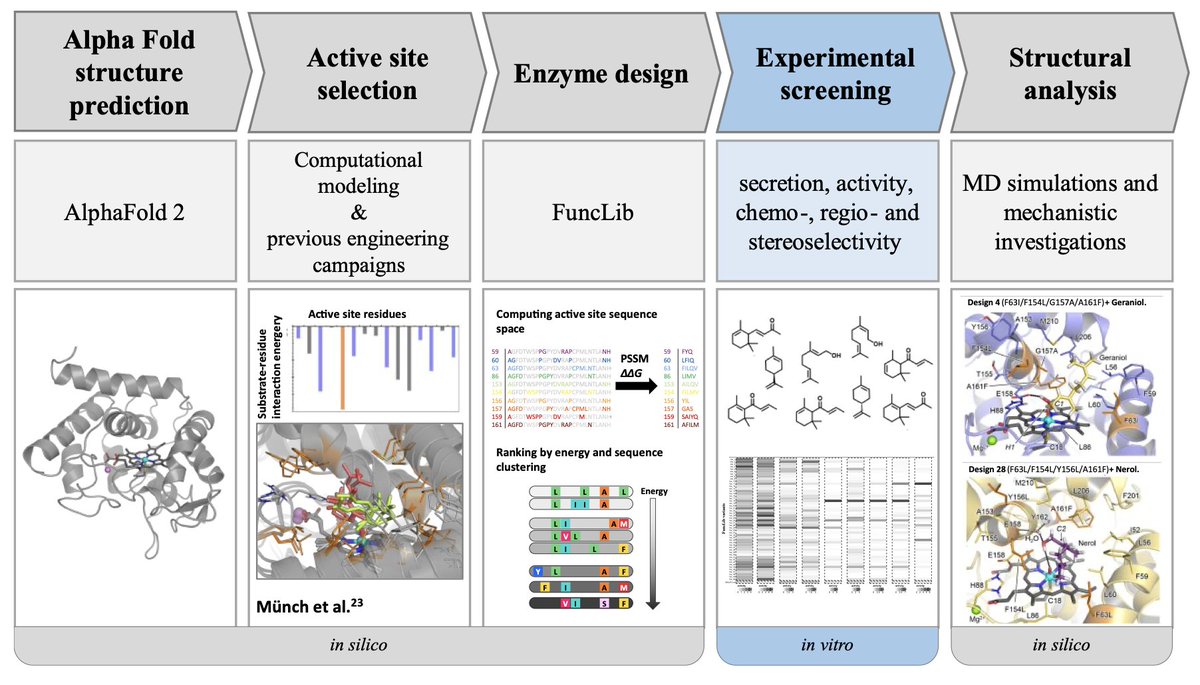
1. This study presents a computational approach to design 50 diverse and selective unspecific peroxygenase (UPO) variants for terpene oxyfunctionalization, all of which were experimentally functional.
2. By leveraging the FuncLib algorithm, the researchers generated multipoint mutations in UPOs that resulted in a wide range of regio-, chemo-, and stereoselective oxyfunctionalization products from terpene substrates.
3. The computational designs demonstrated significant improvements in activity and selectivity compared to the wild type UPO, producing valuable products such as citral, widely used in the fragrance industry and pharmaceutical applications.
4. Notably, the study achieved regioselectivity shifts that favored less chemically reactive positions, which are difficult to achieve through traditional methods, showcasing the power of computational enzyme engineering.
5. This work also highlighted the potential of computationally designed enzymes in green chemistry, as the production of complex oxyfunctionalized terpenes was greatly accelerated, reducing the need for resource-intensive laboratory screening.
6. By using an AlphaFold model and FuncLib, the team successfully bypassed the limitations of traditional enzyme engineering, significantly advancing the development of biocatalysts for industrial applications.
7. The designed UPOs produced new oxyfunctionalization products with remarkable enantioselectivity, such as a dramatic shift to 99% enantiomeric excess for (S)-4-hydroxy-β-ionone, which has high commercial value.
8. This research underscores the utility of computational enzyme design in overcoming challenges like mutational epistasis and lays the groundwork for future enzyme engineering efforts in the field of synthetic biology.
@MarcGBQ
💻Code: https://t.co/EvZ30LmppL
📜Paper: https://t.co/LsmKFLSz9d
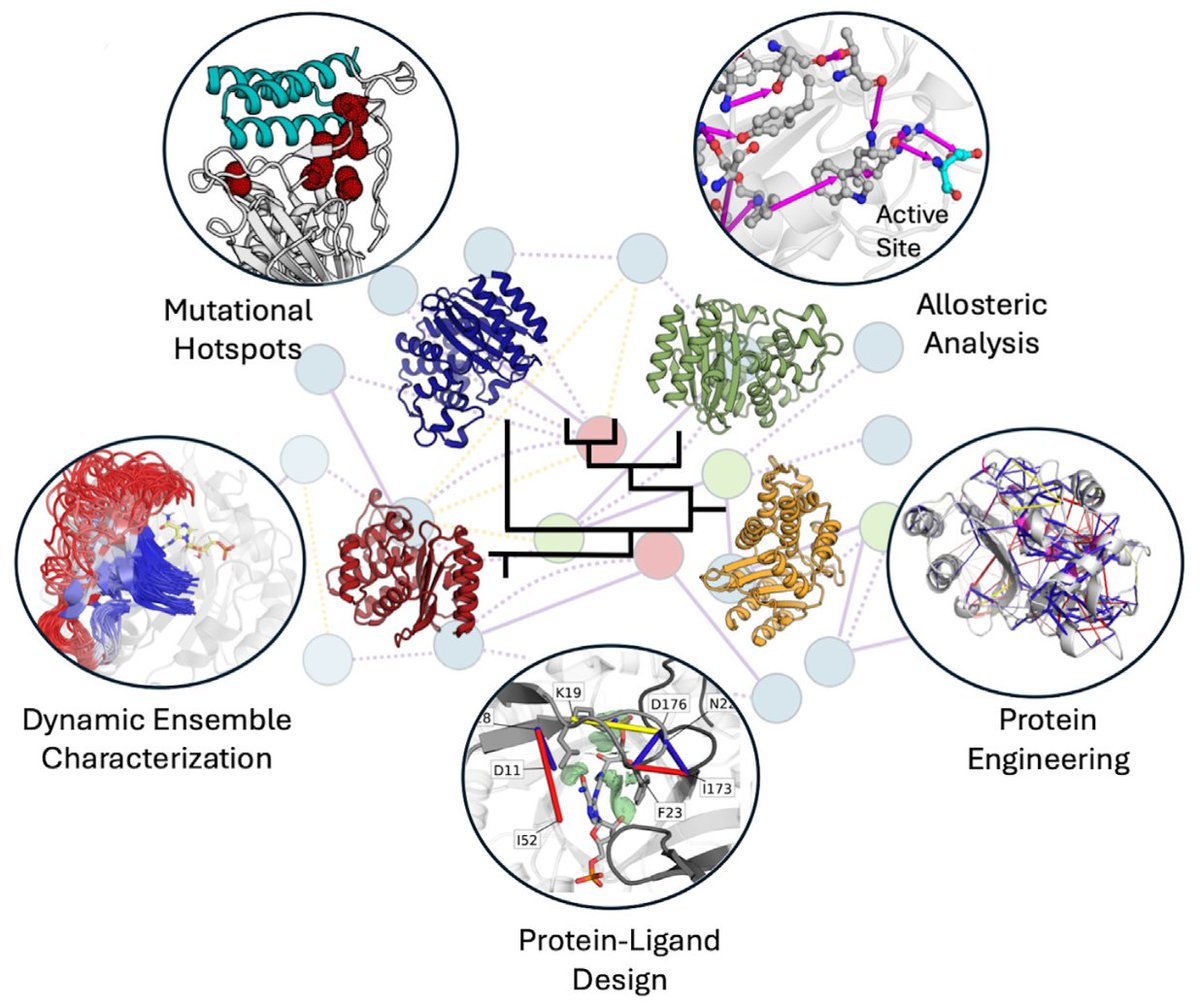
Using residue interaction networks to understand protein function and evolution and to engineer new proteins
- This paper explores the application of residue interaction networks (RINs), which model proteins as graphs, with residues as nodes and interactions as edges. RINs provide a powerful tool for understanding protein structure, function, and dynamics.
- The paper emphasizes the utility of RINs in identifying evolutionarily conserved interaction networks across protein families, guiding both protein engineering and evolutionary studies.
- A standout feature is the ability of RINs to map protein conformational changes, identifying critical residues and mutational hotspots that impact protein folding, stability, and allosteric regulation.
- Meta-RINs allow comparative analysis across protein families, making it possible to trace protein evolutionary trajectories and pinpoint regions essential for protein function, which should be avoided in mutagenesis experiments.
- RIN-based approaches are crucial for engineering new proteins with desired functions, as they help predict the consequences of mutations, thus aiding in rational design efforts.
- The study also discusses advancements in RIN analysis software, highlighting tools like RING, RINmaker, and PyInteraph, which offer new ways to visualize and analyze dynamic interaction networks across protein datasets.
- RINs are being increasingly applied in diverse areas such as drug design, enzyme engineering, and understanding disease mechanisms, making them indispensable for future research in structural biology and biotechnology.
@kamerlinlab@kassonlab@BrDiGeronimo@DYehorova
📜Paper: https://t.co/F5jZBzgEBb
Designing of thermostable proteins with a desired melting temperature
1. Breakthrough in designing thermostable proteins with a targeted melting temperature, boosting applications in industrial and pharmaceutical fields.
2. Developed a regression model using 17,312 non-redundant proteins, achieving a high correlation of 0.89 in temperature prediction accuracy.
3. Combines protein sequence features and embeddings from advanced language models like ProtBert, enhancing predictive performance.
4. Introduced a user-friendly web server and Python package, enabling wider access to protein temperature design tools.
@raghavagps
💻Code: https://t.co/OLjmPDRtdT
📜Paper: https://t.co/n5Q4cd4St5
Modeling protein-small molecule conformational ensembles with ChemNet @UWproteindesign
🚀 New preprint from David Baker!🚀
- ChemNet is a breakthrough deep learning model that accurately predicts the conformational ensembles of small molecules in complex with proteins.
- It outperforms existing methods like AlphaFold and RoseTTAFold by focusing on atomic-level accuracy, generating ensembles rather than single static structures.
- The key innovation is ChemNet’s stochastic nature, which allows it to model conformational heterogeneity, making it ideal for protein-small molecule docking.
- ChemNet generates diverse structures based on random initializations, providing deep insights into the flexibility of molecules at binding sites.
- The model is designed to assess the pre-organization of catalytic sites in enzymes, which is crucial for improving enzyme activity.
- In enzyme design applications, ChemNet-guided designs have achieved impressive success rates, including a retroaldolase with a kcat/KM of 11,000 M-1min-1, the highest pre-deep learning design rate for this reaction.
- The ability to rapidly generate conformational ensembles makes ChemNet a valuable tool for drug discovery and computational enzyme design.
- This system provides a faster, more generalizable alternative to computationally expensive methods like molecular dynamics simulations.
- ChemNet is expected to have broad applications in predicting protein-small molecule interactions, and guiding de novo design of enzymes and drug binders.
@ikalvet@gyurielee@alchemist_an@JustasDauparas
📜Paper: https://t.co/JZG0G5riMK