When you come into biology from CS you scoff at the obscurity of everyone's research, look at all these underpaid postdocs churning out papers on ridiculously niche topics. They don't have the right (startup-adjacent) cultural traits to tackle ambitious goals.
Then...
We're hosting a co-working session for anyone working in Bio+ML in Kendall sq next week. Industry / startups / academia / nonprofits welcome. I will likely do a series of these - if you would like invites to future ones DM me your email!
There's an insane amount happening right now and it feels especially important to break out of silos. Also nice to get something done surrounded by interesting new people
To train better open models, we need predictable scaling.
Delphi is Marin’s first step: we pretrained many small models with one recipe, then extrapolated 300× to predict a 25B-param / 600B-token run with just 0.2% error.
Getting there took some work 🧵
MHCflurry, despite its age, is still somehow the most reliable thing I have for MHC-I presentation prediction.
Anyone want to sponsor the GPU time to train a new major release on updated data?
(@modal? This model gets used quite a bit in vaccine design & cancer immunology)
MHCflurry 2.2.0rc2 is on PyPI:
https://t.co/UCHSxBrtCq
Try it out and let us know if you spot any problems in our transition from TensorFlow to PyTorch
I'm rebuilding AlphaFold2 from scratch in pure PyTorch.
No frameworks on top of PyTorch. No copy-paste from DeepMind's repo. Just nn.Linear, einsum, and the 60-page supplementary paper.
The project is called minAlphaFold2, inspired by Karpathy's minGPT. The idea is simple: AlphaFold2 is one of the most important neural networks ever built, and there should be a version of it that a single person can sit down and read end-to-end in an afternoon.
Where it stands today:
- ~3,500 lines across 9 modules
- Full forward pass works: input embedding → Evoformer → Structure Module → all-atom 3D coordinates
- Every loss function from the paper (FAPE, torsion angles, pLDDT, distogram, structural violations)
- Recycling, templates, extra MSA stack, ensemble averaging — all implemented
- 50 tests passing
- Every module maps 1-to-1 to a numbered algorithm in the AF2 supplement
The Structure Module was the most satisfying part to build. Invariant Point Attention is genuinely beautiful — it does attention in 3D space using local reference frames so the whole thing is SE(3)-equivariant, and the math fits in about 150 lines of PyTorch.
What's next:
- Build the data pipeline (PDB structures + MSA features)
- Write the training loop
- Train on a small set of proteins and see what happens
The repo is public. If you've ever wanted to understand how AlphaFold2 actually works at the level of individual tensor operations, this is meant for you.
Repo: https://t.co/k25vl5th1y
🚀 Just released: Protein Hunter on GitHub!
https://t.co/78TfdNO6i4
Now supports Boltz and Chai with more models coming soon!
Use it to:
1️⃣ Design binders from scratch
2️⃣ Optimize your own designs
🔗 Boltz: https://t.co/7X9id9uDsL
🔗 Chai: https://t.co/P5w2TvoXYI
OpenFold3-preview (OF3p) is out: a sneak peek of our AF3-based structure prediction model. Our aim for OF3 is full AF3-parity for every modality. We now believe we have a clear path towards this goal and are releasing OF3p to enable building in the OF3 ecosystem. More👇
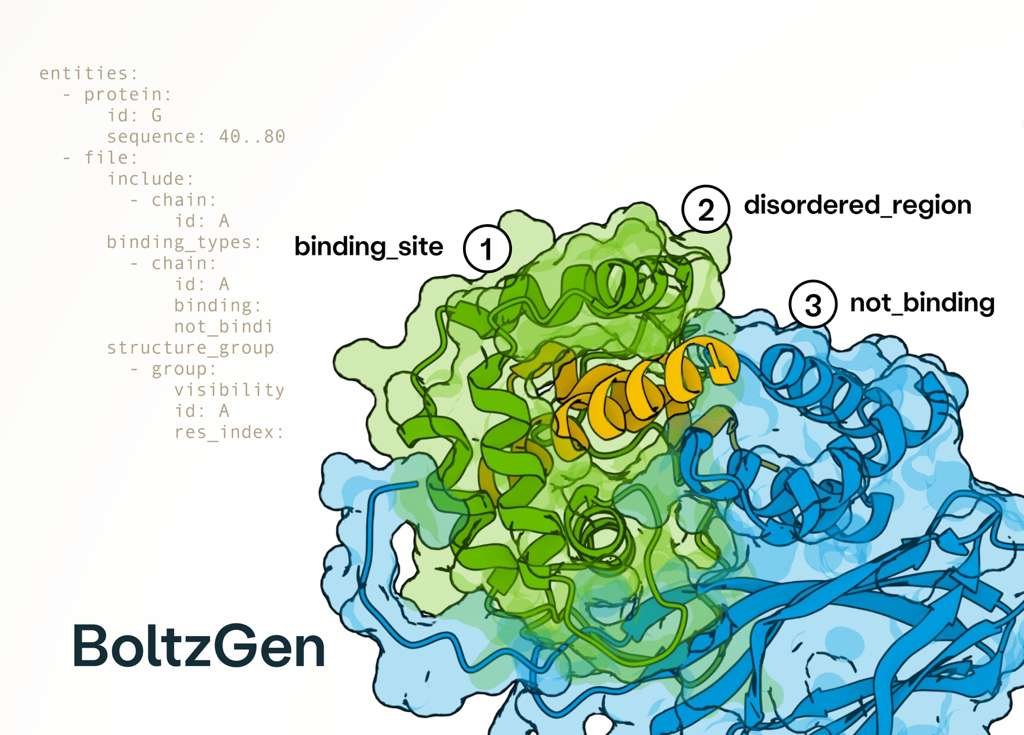
I’ve been testing BoltzGen a bit recently and while I haven’t done any experimental testing yet, the quality of the software is very clear. It installs, runs, logs everything, has tons of options. Very excited to test out the designs irl!
Excited to release BoltzGen which brings SOTA folding performance to binder design! The best part of this project has been collaborating with many leading biologists who tested BoltzGen at an unprecedented scale, showing success on many novel targets and pushing its limits! 🧵..