3D Molecule Generation from Rigid Motifs via SE(3) Flows
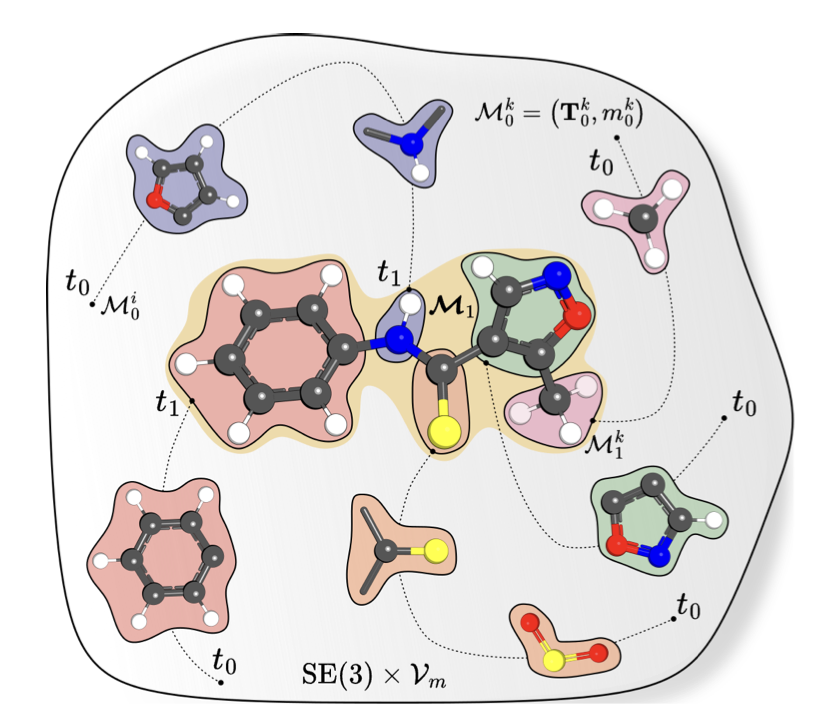
1 MOTIFLOW treats drug-like molecules as sets of rigid-body motifs instead of individual atoms, lifting generation from R³ to the SE(3) manifold and cutting sampling steps 2×–10× versus atom-based diffusion.
2 A data-driven fragmentation canonizes planar rings & fused systems into reusable rigid fragments; motifs are encoded as (P_i, h_i, S_i) where S_i captures discrete rotational symmetries up to order 12, enabling 3.5× representation compression.
3 Joint multimodal flow matching trains a single network to regress SE(3) vector fields for frame poses and CTMC rates for discrete motif identities without autoregressive loops or learned atom-level decoders.
4 On GEOM-DRUGS MOTIFLOW reaches 95 % atom stability and 81 % valid-and-connected molecules with only 100 steps, beating the previous best (87 %, 74 %) that needed 1000 steps.
5 Conditional variants show 95 % exact atom-count satisfaction and 0.86 mean Tanimoto fingerprint similarity on QM9, outperforming strong baselines while using 10× fewer steps.
6 Ablation confirms that keeping planar rings as intact motifs (α = 0.1 % frequency threshold) gives the best stability–coverage trade-off; finer fragmentation oversamples rare substructures and degrades validity.
💻Code: https://t.co/h8d8Sb2aG5
📜Paper: https://t.co/qhSmOMp89N
#DrugDiscovery #3DMoleculeGen #SE3Flow #FragmentBased #MachineLearning #ChemInformatics
Opportunity for #AI scientist in chemistry & pharmaceutical sciences at AstraZeneca (Gothenburg Sweden) closing: 15-Feb-2026 #cheminfomatics#ML#CompChem#EUChemJobs#ChemJobs
https://t.co/X3rOCq12DB
Ultra-large virtual screening discovers agonists of the orphan receptor GPR139, a target for schizophrenia. AlphaFold3 fails to model receptor-ligand complexes for understudied targets! New paper: https://t.co/mmhB0Z2Ssb
@NatureComms
@UU_University
@Scilifelab
SiteMatcher: A Web Server for Structure-Based Drug Design Using Protein–Ligand Interaction Patterns | Journal of Chemical Information and Modeling https://t.co/HVFkIVDA48
A Universal Model for Drug-Receptor Interactions
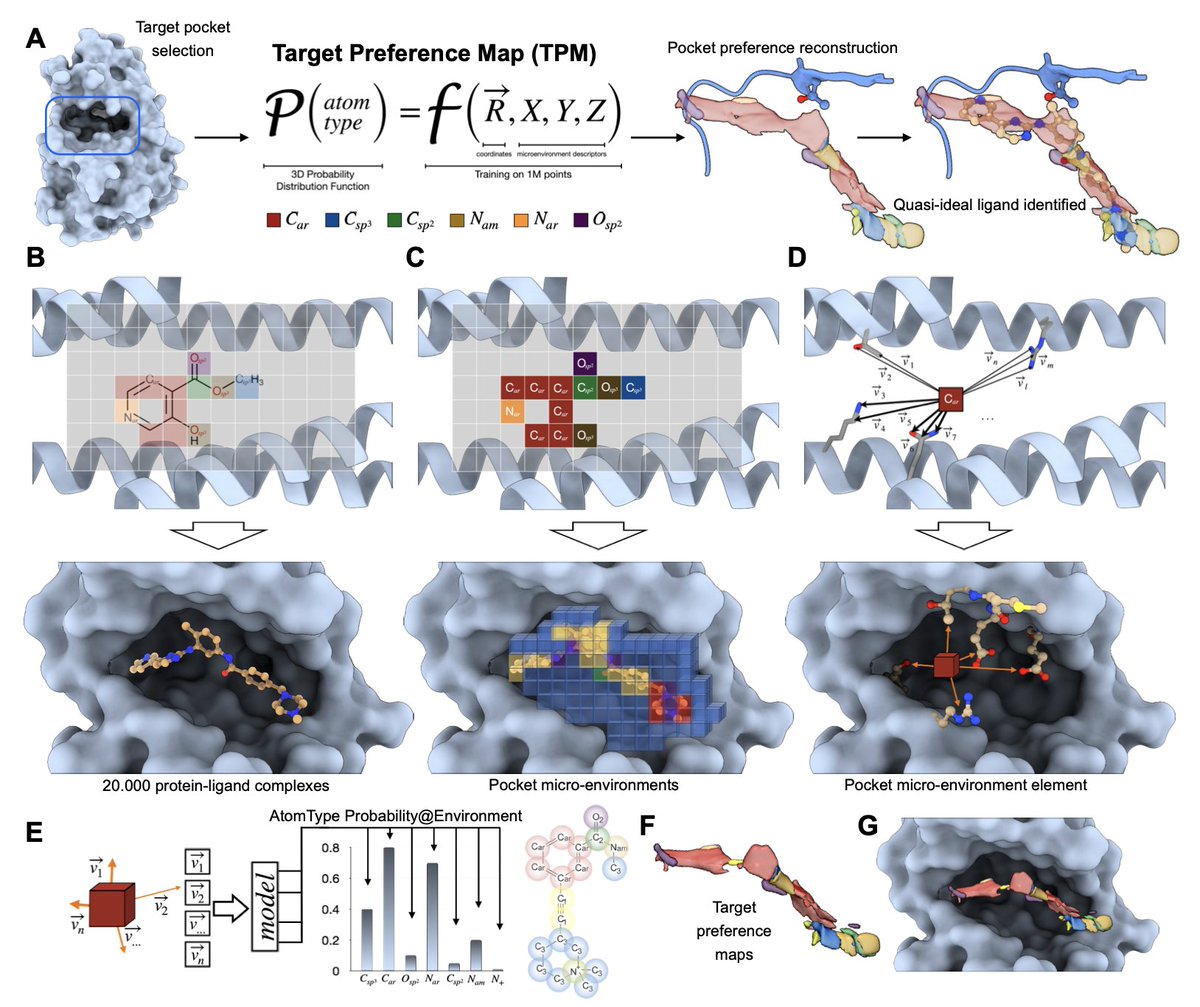
1. A novel study by Menezes et al. introduces a novel machine learning model called Target Preference Maps (TPMs) that aims to revolutionize drug design by predicting optimal drug-receptor interactions. This model generalizes non-bonded interactions, abstracting away chemical identity to focus on spatial interaction fields, thereby capturing the essence of binding events with high accuracy.
2. The TPM model is trained on over 1 million ligand-protein interactions, focusing on local atomic microenvironments rather than entire molecules. This approach minimizes historical bias and enhances the model's ability to generalize interactions, even for structurally diverse compounds. The model's predictions align well with medicinal chemistry principles, leading to improved drug potency.
3. A key innovation of the TPM model is its ability to generate high-resolution maps that visualize spatial binding propensities for individual atom types within a protein pocket. These maps, known as Target Preference Maps, translate the receptor's structure into its drug preference, identifying critical binding features and suggesting optimal modifications for enhanced binding.
4. The study demonstrates the model's effectiveness through both retrospective and prospective validation experiments. In a challenging protein-protein interaction target (PEX14-PEX5 interface), the TPM model guided the synthesis of new inhibitors that showed significant improvements in potency, highlighting the model's practical value in drug design.
5. The TPM model also shows remarkable sensitivity to minute structural details, such as point mutations and cofactor interactions. For example, it accurately predicts the presence of bridging water molecules and the optimal placement of functional groups near metal ions, showcasing its potential for precision medicine and complex target optimization.
6. Limitations of the current TPM framework include its agnosticism to chemical connectivity and its reliance on static, experimentally resolved structures. Future work could address these limitations by incorporating molecular dynamics and energetic modeling to capture receptor dynamics and improve the model's predictive scope.
📜Paper: https://t.co/P1kK28aQnL
#DrugDesign #MachineLearning #ComputationalBiology #ProteinLigandInteractions #PrecisionMedicine
Fast and Accurate Prediction of Tautomer Ratios in Aqueous Solution via a Siamese Neural Network | Journal of Chemical Theory and Computation https://t.co/s6boDwdLYo
Accelerating the Hit-To-Lead Optimization of a SARS-CoV-2 Mpro Inhibitor Series by Combining High-Throughput Medicinal Chemistry and Computational Simulations | Journal of Medicinal Chemistry https://t.co/RV52vCfKpC