Extremely excited to announce LigandForge 🧬⚡
Generate high-quality peptides at over 10,000x - 1M the speed of state-of-the-art methods like Bindcraft and Boltzgen. Predict binding affinity with 83% correlation to experimental binding data. 150 protein targets benchmarked.
The biggest-selling drug on the planet last year was a peptide. Semaglutide, the molecule inside Ozempic and Wegovy, is a chain of just 31 amino acids. It generated roughly $33 billion in revenue for Novo Nordisk in 2025. One molecule. The entire peptide drug market crossed the $50 billion mark.
Finding the right peptide is where all the money burns. For each disease protein, you need to design a peptide that binds tightly enough actually to work. Think of it like making a custom key for a lock, except each position on the key can take 20 possible shapes, and even a short 10-position peptide can have over 10 trillion possible combinations per target.
The two best AI tools for this, BindCraft and BoltzGen, work by first predicting a peptide's 3D shape, then checking whether it sticks. That two-step process generates one candidate every few seconds to a few minutes. A whole day might get you a few hundred designs.
LigandForge skips the shape-prediction step entirely. It learns the physics of molecular interactions and generates sequences directly from the shape of the target protein’s docking site. No iteration, no structure prediction during generation. Over 700 peptide sequences per second on a single GPU. That’s 10,000x faster than BoltzGen, over 1,000,000x faster than BindCraft.
Speed means nothing if the peptides are garbage, though. So they tested it on five targets that have historically been difficult to bind: TNF-alpha, the target behind the rheumatoid arthritis blockbuster Humira. PD-L1 is the immune checkpoint that cancer immunotherapy drugs like Tecentriq block. VEGF-A, the target for cancer drug Avastin. HER2, breast cancer drug Herceptin’s target. And IL-7R-alpha.
LigandForge generated 150,000 candidates across all five in 3.4 minutes and produced tightly binding candidates (predicted binding strength in the low nanomolar range, where real drugs operate) against all five. BoltzGen hit 1 out of 5. BindCraft hit 0.
A 2020 JAMA study pegged the median cost of bringing a single drug to market at $985 million. The early discovery phase, where you’re searching for molecules that bind to your target, can take 1 to 6 years. A tool that searches the same space a million times faster changes how many disease targets a lab can afford to go after at once.
Global catastrophe since many incredible programs are not currently being funded. Parasites don’t carry passports. Diseases spread across countries, being able to survey & address these pathogens in one part of the world benefits public health everywhere
https://t.co/UYUj0Bx6TO
How a self driving car motivated us to build a new “universal diagnostics” platform. The arc of technology is unpredictable at best.
https://t.co/AVQxreaj8L
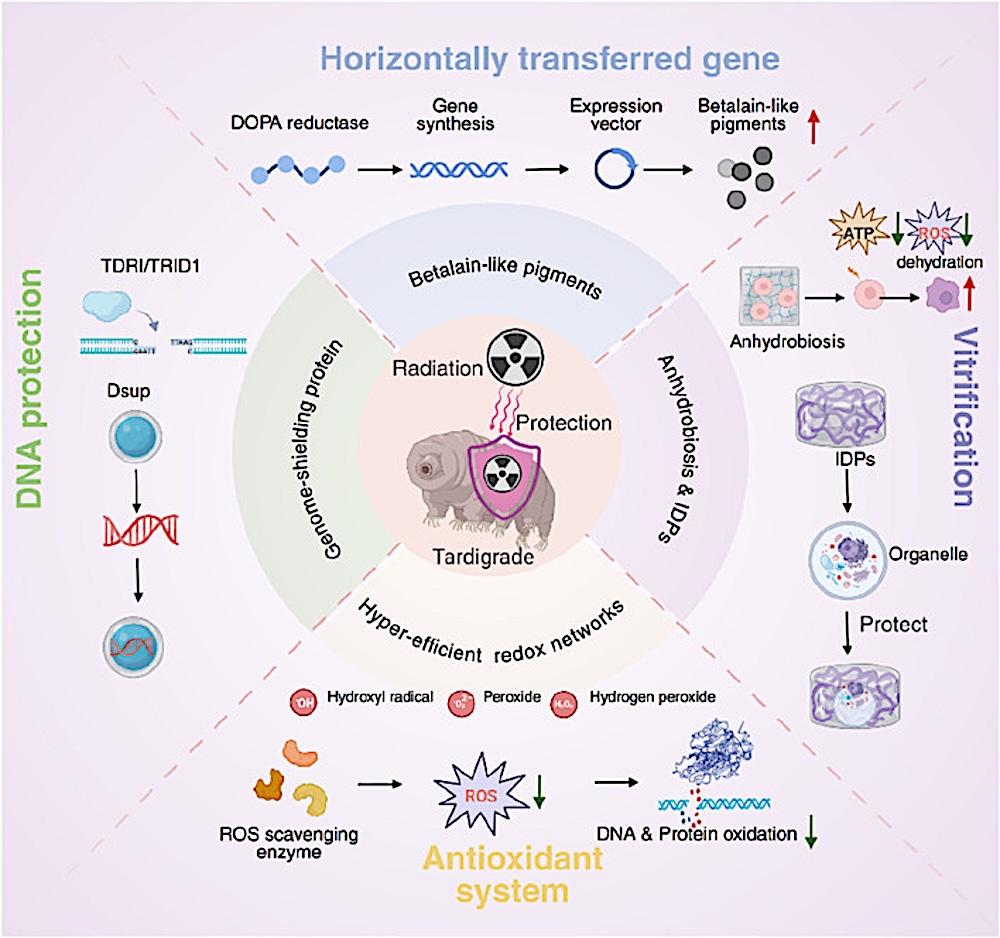
Radioprotection Redefined: Drug Discovery At The Intersection Of Tardigrade Biology And Translational Pharmacology
https://t.co/SPV5oLjih5 #astrobiology#extremophile#Tardigrade @tardigradopedia
Today marks exactly 10 years of start of an incredible journey of @TeamFoldscope - we have grown older and wiser and so has our curiosity. To everyone who has supported curiosity over this last decade - thank you! 🙏🏾 see https://t.co/eFcC8ezVG0 and dive into the world of small.
If one drops identification with one’s own body and mind, which are accumulations of food and impressions, then one shall know stillness within. That is all it takes to become meditative. #SadhguruQuotes
Life knows how to keep going - even if it’s freezing -15 degrees C 🥶 outside. AGU just covered our discovery of ultra resistant ice diatoms gliding into a world record! Enjoy the story here:
https://t.co/azKZQcd68A
Way too many people think that AlphaFold "solved" ML for proteins.
It didn't.
It did revolutionize protein structure prediction, but that’s just one part of a much bigger puzzle.
This is Part 1 of a series on what AlphaFold did (and didn’t) solve—and what comes next. ⬇️
Lots of hype around multimodal FMs, virtual cells (and labs?), all-atom design...I really think core algorithms (not just scale/integration) will solve the next problems in AIxBio. Take Transition Path Sampling: models transitions for dynamics, optimization, and cell fate. 👇
Even as a sequence-first person, these are some papers TPS papers I've been excited about from the past few conferences!
-Doob's Lagrangian (NeurIPS '24): you can now model state transitions like cell differentiation or conformational shifts using time-symmetric stochastic control. Basically, it learns optimal path ensembles without predefined dynamics!
https://t.co/VFDvkHU8Dr
-TPS-DPS (ICLR '25): want to simulate how a protein folds or a molecule binds, without defining reaction coordinates? This learns likely transition paths directly from data using diffusion models (see the GIF below)!
https://t.co/UrgXUjf5UU
-TPS with Onsager-Machlup Functional (ICML '25): One of my favorites: samples the most probable stochastic trajectories by minimizing action under the OM functional. Ideal for capturing noisy yet structured dynamics: folding funnels or switching paths. Physics + generative models = 🔥
https://t.co/PKdOsUgPtW
If you're just paying attention to just the hype-y models, you'll miss some very good stuff! 🫣 So many exciting translational steps to take with these new methods!
If you are in Bangalore or surrounding area on 6th Sept - it would be fantastic to see you. We are starting a broader conversation on scaling up https://t.co/JZyDjXNNB6 - if you want to teach these principles in your classroom - pls reach out here.. @TeamFoldscope
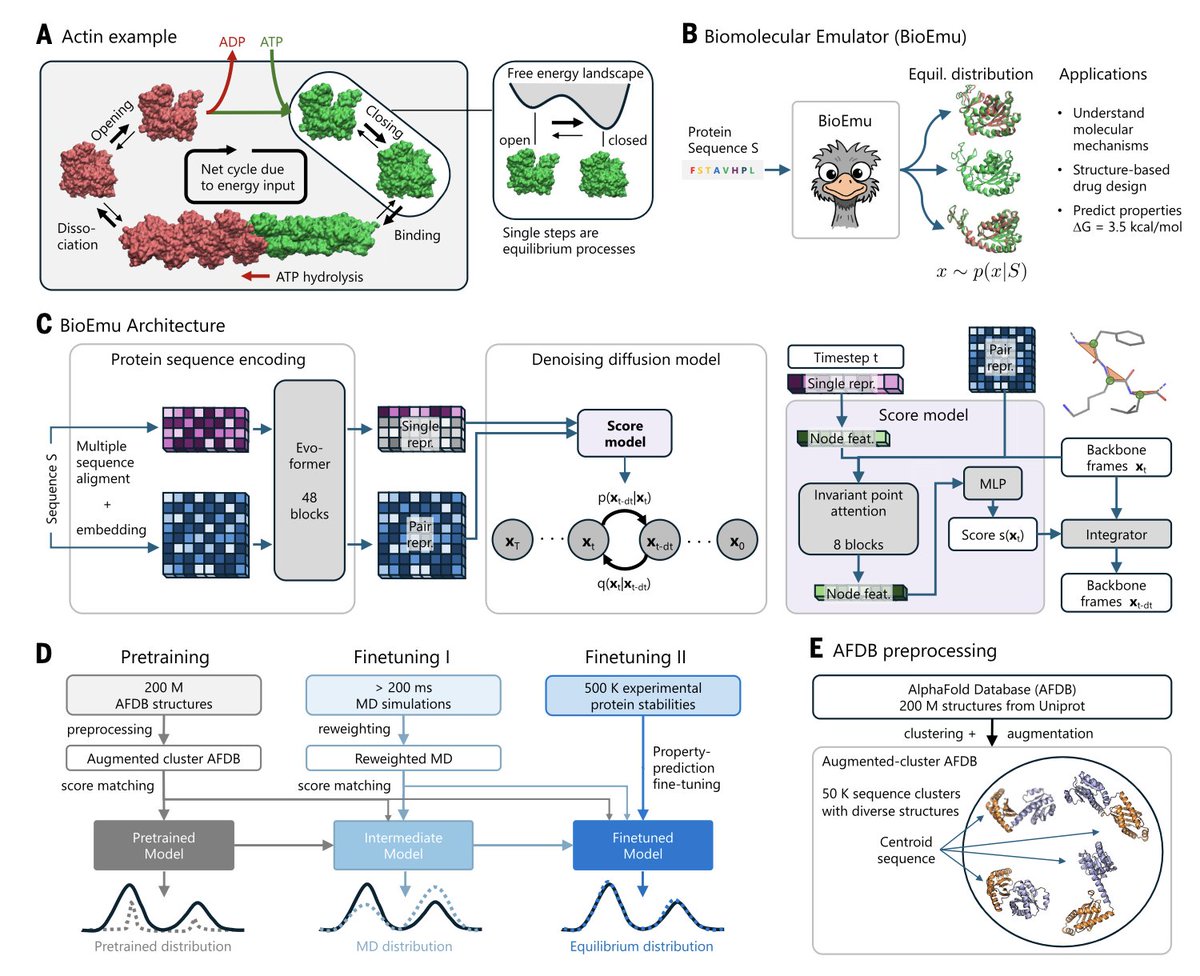
Scalable Emulation of Protein Equilibrium Ensembles with Generative Deep Learning @ScienceMagazine
1. A groundbreaking study introduces BioEmu, a deep learning system that can rapidly generate protein structure ensembles, capturing dynamic conformational changes and predicting protein stability with high accuracy. This innovation could revolutionize drug discovery and biotechnology by providing a scalable way to understand protein function.
2. BioEmu leverages a combination of AlphaFold for sequence encoding and a diffusion model for structure generation. It was trained on over 200 milliseconds of molecular dynamics simulations and 500,000 experimental protein stabilities, demonstrating its ability to predict known conformational changes and emulate equilibrium distributions with remarkable speed and accuracy.
3. The model's ability to predict protein stability is particularly noteworthy. BioEmu achieved a mean absolute error of less than 0.9 kcal/mol and a Spearman correlation coefficient of approximately 0.6 for folding free energies, showing its potential for practical applications in predicting the stability of mutant proteins.
4. BioEmu's architecture and training approach are designed to handle the complexity of protein dynamics. It can sample thousands of independent protein structures per hour on a single GPU, making it orders of magnitude faster than traditional molecular dynamics simulations. This efficiency opens new avenues for high-throughput protein analysis.
5. The study also highlights BioEmu's potential for drug discovery by identifying binding pockets and allosteric mechanisms. It can generate ensembles for dynamical protein design, providing insights into how proteins work at a molecular scale and paving the way for the development of new therapeutic strategies.
6. The authors emphasize that BioEmu is not intended to replace molecular dynamics simulations but rather to complement them. It can be used to generate initial ensembles for further refinement with MD, offering a cost-effective and scalable alternative for exploring protein function.
📜Paper: https://t.co/TE6tSnN6gg
#BioEmu #ProteinDynamics #DeepLearning #DrugDiscovery #Biotechnology #ProteinStability
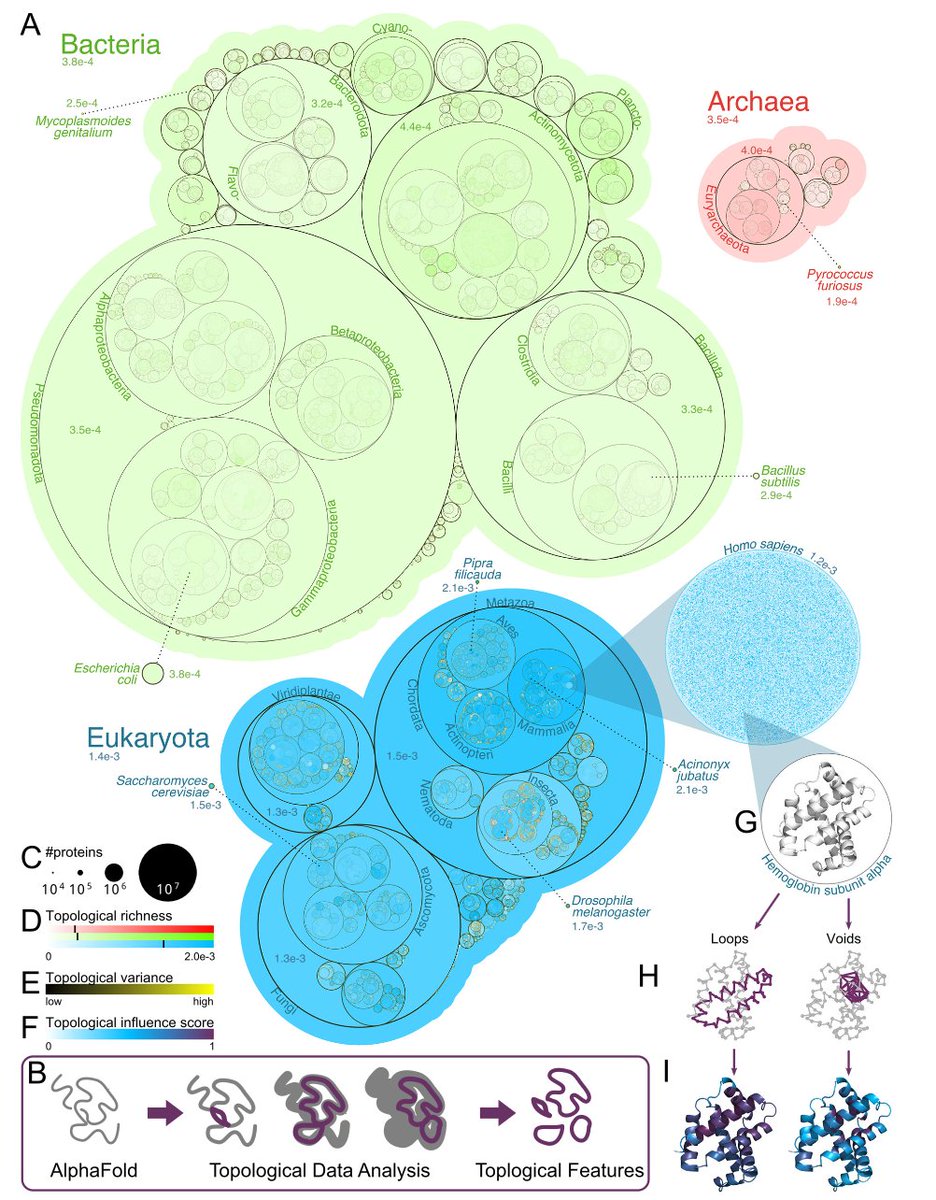
The Topological Properties of the Protein Universe @NatureComms
1. This novel study leverages topology—a branch of mathematics—to analyze the structural intricacies of over 214 million proteins predicted by AlphaFold2. By focusing on qualitative spatial features, it offers a novel lens to understand protein structure-function relationships and evolutionary patterns.
2. The researchers developed a pipeline using persistent homology to map the topological features of the entire protein universe. This approach identifies key organizing principles, such as domain architecture and binding sites, and reveals differences between proteins from mesophiles and thermophiles.
3. A significant finding is that eukaryotic proteins, especially those from multicellular organisms, exhibit higher topological complexity compared to bacterial and archaeal proteins. This complexity may be linked to the intricate regulatory mechanisms required for multicellular life.
4. The study demonstrates that topological analysis can detect regions in proteins that are enriched for disease-associated mutations. This suggests that topological features could serve as biomarkers for structural stability and potential targets for therapeutic interventions.
5. The researchers also compared thermophilic and mesophilic proteins, finding that thermophilic proteins have smaller and more compact voids. This topological difference may reflect adaptations to high-temperature environments and provide insights into protein stability.
6. The study provides a comprehensive topological “tour guide” to the AlphaFold2 database, making the data freely available online. This resource enables researchers to explore the vast protein universe and uncover new insights into protein structure and function.
7. The innovative use of topological data analysis (TDA) in this study highlights its potential for large-scale protein structure analysis. By integrating TDA with existing bioinformatics tools, researchers can gain deeper insights into the molecular machinery of life.
📜Paper: https://t.co/iwYWpcbvdn
#ProteinStructure #Topology #AlphaFold2 #Bioinformatics #ProteinUniverse #StructuralBiology #EvolutionaryBiology