AlphaGenome is out in @nature today along with model weights! 🧬
📄 Paper: https://t.co/1fHzSPiY1x
💻 Weights: https://t.co/z6JWLT4Mpv
Getting here wasn’t a straight path. We sat down @googledeepmind to discuss the story behind the model, paper & API: https://t.co/cT8CiXfnxQ
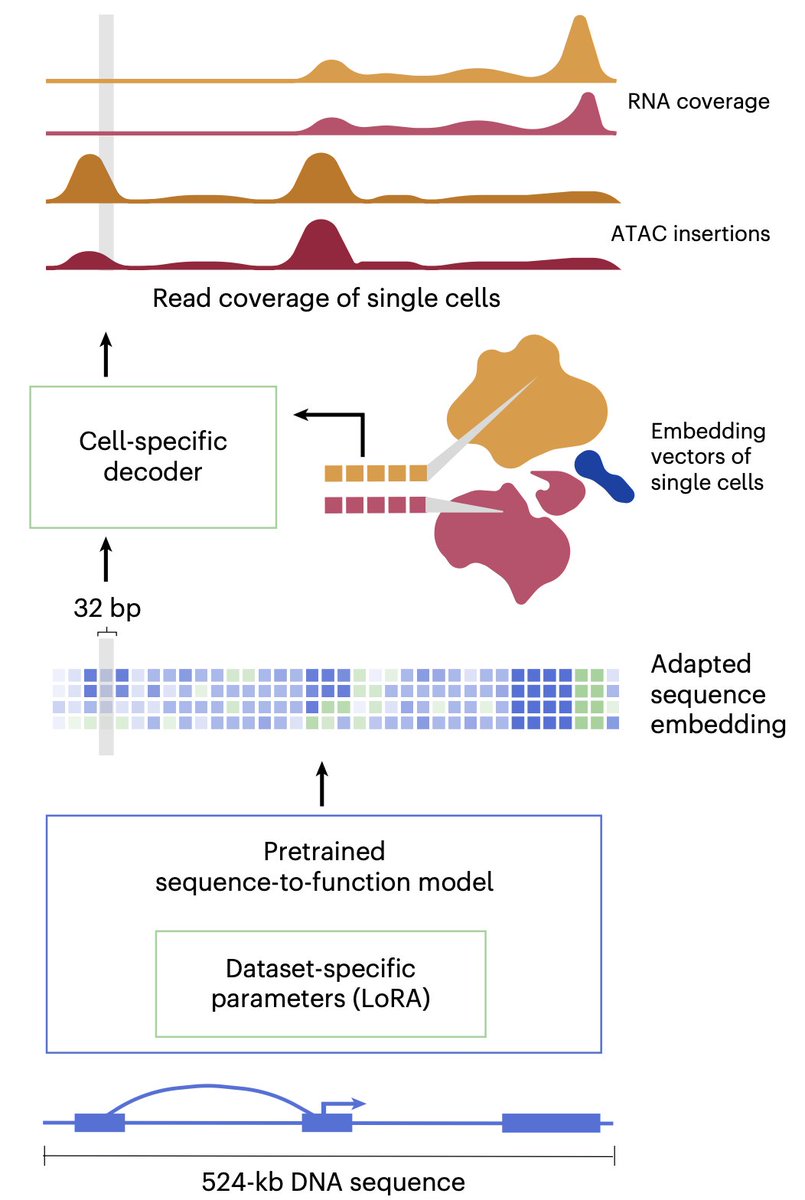
I'm excited that our paper, "scooby: modeling multimodal genomic profiles from DNA sequence at single-cell resolution," has been published in Nature Methods! scooby is a new deep-learning framework to understand how DNA sequence shapes gene expression in individual, single cells.
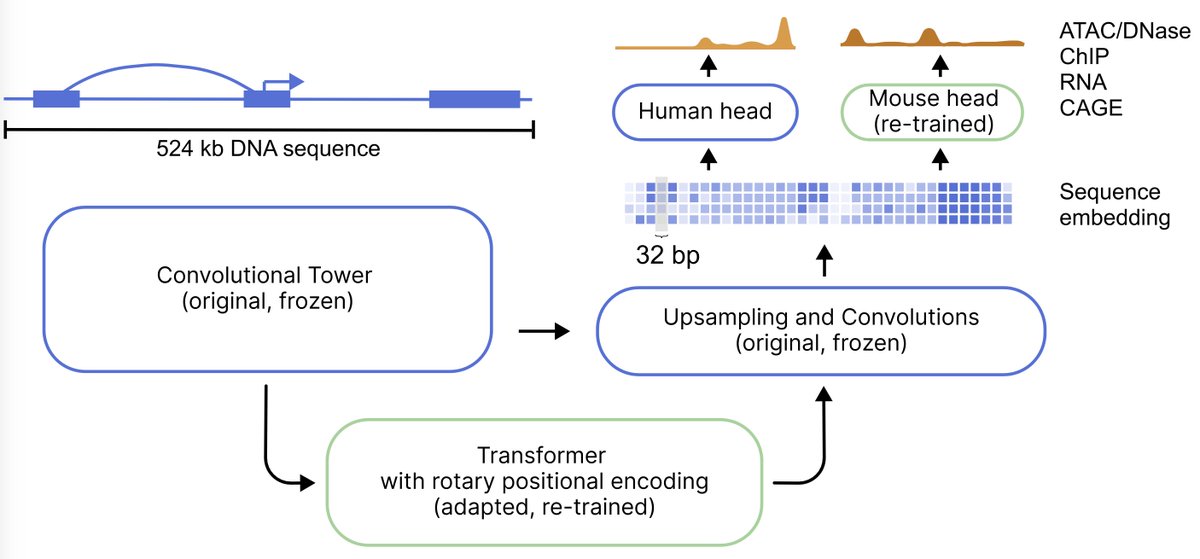
Happy to share that Flashzoi is now published!
We enhanced Borzoi with RoPE & FlashAttention for >3x faster training/inference & 2.4x reduction in memory usage.
This brings large-scale genomic analysis and fine-tuning within reach of academic budgets.
📄: https://t.co/Llyt6k0YLK
Excited to share #AlphaGenome, a start of our AlphaGenome named journey to decipher the regulatory genome! The model matches or exceeds top-performing external models on 24 out of 26 variant evaluations, across a wide range of biological modalities.1/6
Introducing Flashzoi⚡! We’ve upgraded the Borzoi model with rotary pos. encodings and FlashAttention, resulting in a 3x speedup with similar or better accuracy for faster variant effect prediction or model development, and more efficient genomic analysis https://t.co/yIGRWHUS3b
Excited to share our preprint: Efficient Inference, Training, and Fine-tuning of Protein Language Models! ⚡We introduce advanced techniques to make large protein models accessible to more researchers. Read it here: https://t.co/qkeebt8gbO
Information leakage due to homology: A pervasive and non-trivial issue in bioinformatics. Excited to hear about @muntakim_rafi's insights and recommendations for sequence-based modeling on Wednesday at the Kipoi seminar.
Excited to present “scooby”, which models multi-omic profiles (scRNA-seq coverage & scATAC-seq insertions) directly from 500 kb DNA sequence at single-cell resolution. This was a fantastic collaboration co-led with @thisisjohahi.
https://t.co/UEG56QFFwt
@sokrypton@pedrotomazsilva@anshulkundaje I see, but my point there was more that the DNA seqs we look at generally aren‘t one „unit“ like one protein, but a collection of elements that need not be linked. If you have high within-element dependency, then centering will distribute this signal intra-element or?
@sokrypton@pedrotomazsilva@anshulkundaje Also we often see broad interactions that represent logically linked elements. E.g. in fig3B, the splice sites interacts with the intron and CDS. Centering this would enforce a compensating negative interaction between splice-site and intergenic region that I find hard to justify
@sokrypton@pedrotomazsilva@anshulkundaje One of the things we want the model to do is tell us which positions are even doing something - anything - at all. And we show that the row-sum seems a decent proxy of this, sometimes quite a bit better than „traditional“ conservation.
@anshulkundaje@sokrypton@pedrotomazsilva With that caveat, on the lac element there are some *hints* of it maybe picking up on the link between binding sites. But more concrete answer requires a good method to inspect content of final embeddings I think
@anshulkundaje@sokrypton@pedrotomazsilva Hard to say. Evo only gets context from 5‘, so must see a few bases of an element before it „knows“ what its seeing. For short & mobile motifs, its quite likely that it only knows it has seen a motif ex-post, so it might be reflected in final embedding, but not in local nuc probs
@sokrypton@pedrotomazsilva Cool! For us Evo generally gets all tRNA hairpins in E. coli - not just local. IIRC it even gets the weird selenocysteine tRNA mostly right. I think Evo had some bug in their huggingface model for a while, this might explain the difference.
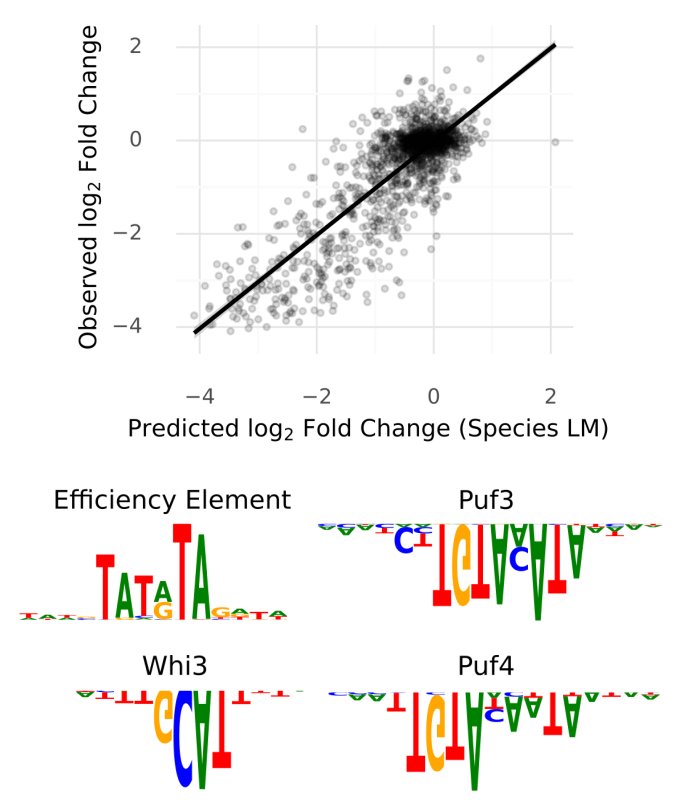
Have you ever wondered what the genome looks like through the eyes of a DNA language model? In our newest preprint we use DNA LMs to study nucleotide dependencies in the genome, revealing functional elements, characterizing variants and evaluating DNA LMs https://t.co/oJaXLjxc95
@vagar112 @NatureGenet I think the fact that now people are seriously asking questions like „can DL models predict personalized expression without retraining“ implicitly demonstrates the advance Enformer was. Even if its not there yet, it is at least conceivable - I dont think it really was before
Cracking the regulatory code: We have genomes for 1000s of species, but ENCODE only for 2 – what do we do? Natural language models have shown that syntax and semantics can be learned from text alone. Can we do the same for genomes?⬇️
https://t.co/MIFqNVfAlC
In all these tasks, providing the model with species information was crucial. We expect that species-aware DNA language models leveraging massive sequencing projects will prove a powerful tool to investigate understudied species.
Finally, our models have learned representations that can boost supervised learning. In fact, we outperform SOTA on gene expression and mRNA half-life tasks using simple regression on the last layers embeddings.