AlphaGenome is out in @nature today along with model weights! 🧬
📄 Paper: https://t.co/1fHzSPiY1x
💻 Weights: https://t.co/z6JWLT4Mpv
Getting here wasn’t a straight path. We sat down @googledeepmind to discuss the story behind the model, paper & API: https://t.co/cT8CiXfnxQ
Agents for computational biology are advancing rapidly, but evals are lagging. Current benchmarks can be overly prescriptive. Full analysis vignettes are hard to verify. We introduce CompBioBench: 100 diverse, challenging, verifiable tasks + benchmark general-purpose agents. 1/9
Check out a vetted Pytorch port of AlphaGenome by GenomicsxAI collaborative team. See QT thread (with links to code & blogpost). Various fine tuning modules + tutorials coming next. Community building announcements coming soon as well. Follow blog for latest updates.
We built a lightweight wrapper to fine-tune AlphaGenome on genome-wide assays (RNA-seq, ATAC-seq, ChIP-seq) and MPRAs.
To keep it accessible, we included blog posts, markdown guides, and notebooks, so you can use whichever format works best for you!
Links in the thread:
We’ve been working with AlphaGenome for a while — today we’re sharing two updates:
• alphagenome-ft: a lightweight structured fine-tuning wrapper
• MPRA fine-tuning results with SOTA performance
Adapting AlphaGenome's JAX/Haiku stack to new assays isn’t trivial. 1/8
Excited to share new finetuning scripts for AlphaGenome in JAX! This was in collaboration with @anshulkundaje group.
As part of the initial release, we provide lightweight wrapper to finetune AlphaGenome on functional genomics (i.e. bigwig) tracks.
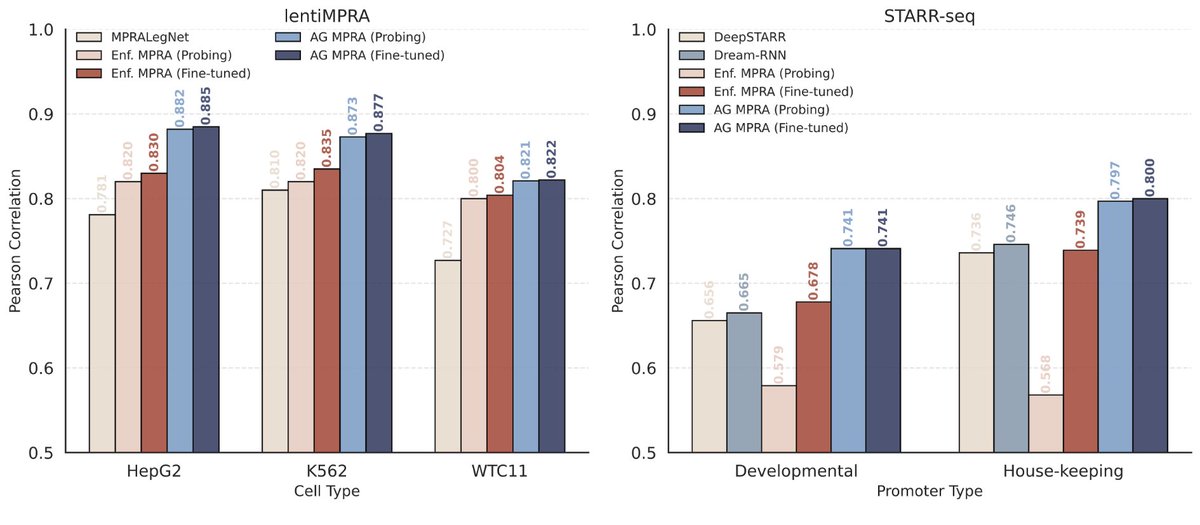
Can AlphaGenome, which considers 1 megabase DNA as input, be finetuned on short DNA sequences from MPRAs (~200bp)?
@Al_Murphy_ shows this approach can lead to state-of-the-art performance on lentiMPRA!
Check out the companion blog post:
https://t.co/JWFWUezGGV
@TuXinming@anna_spiro@ChikinaLab@sara_mostafavi@anshulkundaje Nice work. Worth also pointing out that individual expression prediction with seq2func models is much more difficult bc you need to correctly predict the effects of *all* variants in LD. The errors compound and a single wrong prediction can flip the sign.
AlphaGenome paper and models are out today! https://t.co/WHejipHfu7. We have seen great community engagement since we released the API. We hope to open more use cases with the model weights! https://t.co/TUfKPeOYyY
We updated the supplementary note with some very interesting case studies. I encourage everyone using AlphaGenome for non-coding variant interpretation to read it.
It was a huge team effort, I’m very proud of all of our co-authors @googledeepmind!
Finally, huge congratulations to the amazing team behind this work! 👏
And thank you @demishassabis & @pushmeet for fostering such a fantastic research environment @googledeepmind for science.
AlphaGenome is out in @nature today along with model weights! 🧬
📄 Paper: https://t.co/1fHzSPiY1x
💻 Weights: https://t.co/z6JWLT4Mpv
Getting here wasn’t a straight path. We sat down @googledeepmind to discuss the story behind the model, paper & API: https://t.co/cT8CiXfnxQ
Highlighting two manuscript additions:
1. Data ablation (Supp. Fig 12): RNA-seq + ATAC/DNase proved essential for key evals. h/t @anshulkundaje for the suggestion!
2. Variant Analysis: We found evidence to upgrade a 3’ UTR VUS from Ellingford et al 2022 to "likely pathogenic."
@SayanGhosal94@anshulkundaje Variants that alter a single vs multiple tracks has nothing to do with the result you are seeing. It's whether the model has seen individual level data or not.
As i said, to test actual generalisation you need to test on variants on held out chromosome in this case.
@SayanGhosal94@anshulkundaje Correct evaluation for casual effects is to look at fine-mapped eQTLs with high pip on a completely held out chromosome if your model is training on individual data.
@SayanGhosal94@anshulkundaje The vast majority of eQTLs are spurious associations due to LD. Models that try to capture the causal effects of variants on expression (e.g. Borzoi, AlphaGenome) will therefore have near 0 correlation (as desired). Looks like your model is learning these spurious associations.
My Journal Club on Enformer, an AI model that predicts gene expression from DNA, is out in @NatureRevGenet
This is a high-level look at how Enformer advanced seq2fun AI, and the challenges that remain in modeling gene regulation.
(Easy 5 min read)
🔗 https://t.co/VoFlE2xUJT
@anshulkundaje@i000@jboysen0@kjaganatha For AlphaGenome, the forum https://t.co/aQFON77R2j could be a good place. It would be interesting if there was a way to programmatically/systematically define these (input+expected output) so that they could be used for future evaluations.
Thrilled to introduce AlphaGenome, our new DNA sequence model now available via our AlphaGenome API. Really excited to see how the scientific community uses AlphaGenome’s predictions to understand genome function, drive biological discoveries, develop new treatments, and more...
Excited to share #AlphaGenome, a start of our AlphaGenome named journey to decipher the regulatory genome! The model matches or exceeds top-performing external models on 24 out of 26 variant evaluations, across a wide range of biological modalities.1/6