Excited to share our latest paper led by Renata Goncalves @RenataG81952725: CoQ imbalance drives reverse electron transport to disrupt liver metabolism.
https://t.co/svgsVGKCfd
We uncover how altered CoQ redox balance triggers RET and rewires hepatic metabolism to offer critical insights into a decades old question. Meticulously mapping each one of the 11 sites of the mitochondrial electron transport chain in the liver showed site IQ as the sole source of excess mitochondrial ROS, produced via reverse electron transport. This defect is driven by CoQ imbalance in obesity, disrupts glucose metabolism.
These findings may have implications for many diseases and open up new treatment possibilities where broad antioxidants have failed to generate therapeutic benefit. Our findings may also offer a potential mechanism and solution for the increased diabetes risk seen in a small fraction of statin users.
Huge congratulations to Renata Goncalves @RenataG81952725 for leading this work and for her immense dedication and patience to bring it to a conclusion. Congratulations to all contributors from the @hotamisligil lab and our amazing collaborators Isabel Graupera @igraupe and Shawn Burgess @Isotopomer
Curious? The 93-page-long peer review file is full of useful information and insightful discussions.
Thrilled to share that our latest study is now published in @Nature!
Grateful to our amazing collaborators at @gshlab and @MET_HSPH.
https://t.co/hknvlAj6Dv
https://t.co/OUwnI6rLV8
More insights from our study published today in @Nature: We uncover how mitochondrial dysfunction in the liver contributed to drive insulin resistance in obesity. Read the @HarvardChanSPH press release 👉 https://t.co/iL9vDTajcp
Using idc rates to optimize research deserves study and modification. But the approach just taken was designed not to improve the process, but to harm institutions, researchers and biomed research. Many supporting this action seem ignorant about the US biomed research ecosystem.
Our latest paper is out! In collaboration with @DrAnneCarpenter, we conducted a systematic high-content screen to understand the role of protein mislocalization in diverse human disorders. 1/ https://t.co/pQzVx0hOrC
Let’s play a little game.
Let’s say that you’re the CSO at a cancer pharma company, and you have to choose a target to go after.
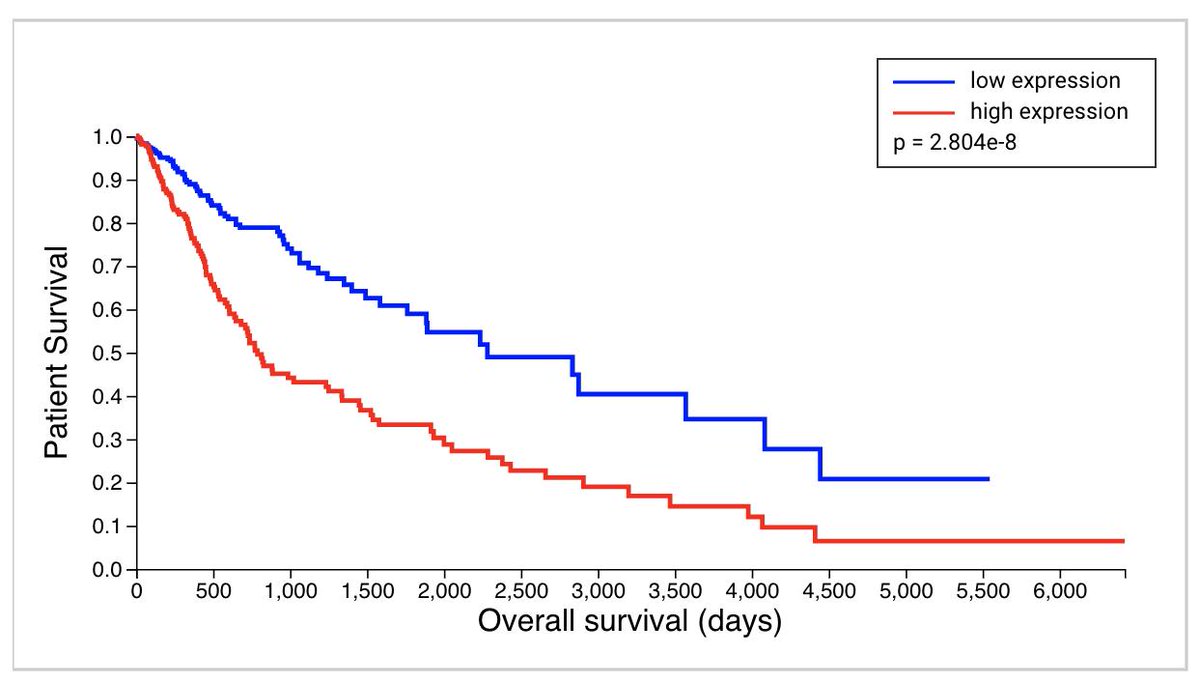
Here’s a gene – high expression is associated with poor prognosis in brain cancer. Looks like a good candidate for an inhibitor right?
@gangxue0502 Thank you! We just wrote our own python functions to visualize the splice forms. The problem with all the existing solutions was that we were only interested in the protein coding regions for our study. All the code is here: https://t.co/yU5JNeUwEx
Prospective power analysis is hilarious because your replace an arbitrary guess of sample size with an arbitrary guess of the effect size you’ve yet to measure, do a touch of arithmetic, and suddenly it’s considered rigorous
@jankosinski@GoogleDeepMind Thank you Jan for the intensive testing and interesting findings! For the templates, it is the same cutoff as mentioned in the paper, i.e. 2021-09-30. We will add this to the Server FAQ for clarity and will let users know if we change this date
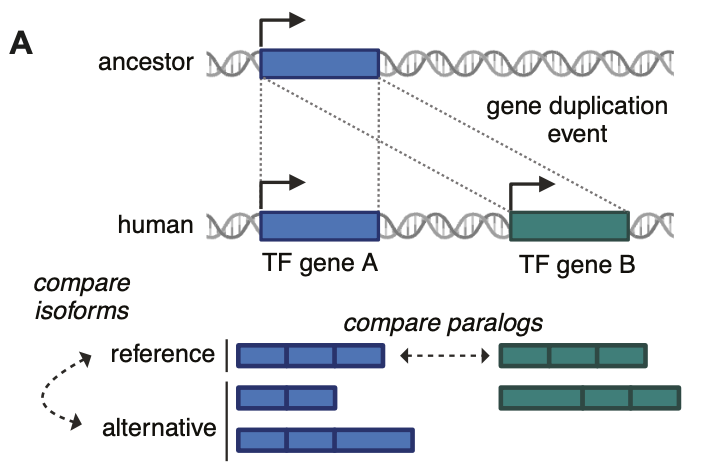
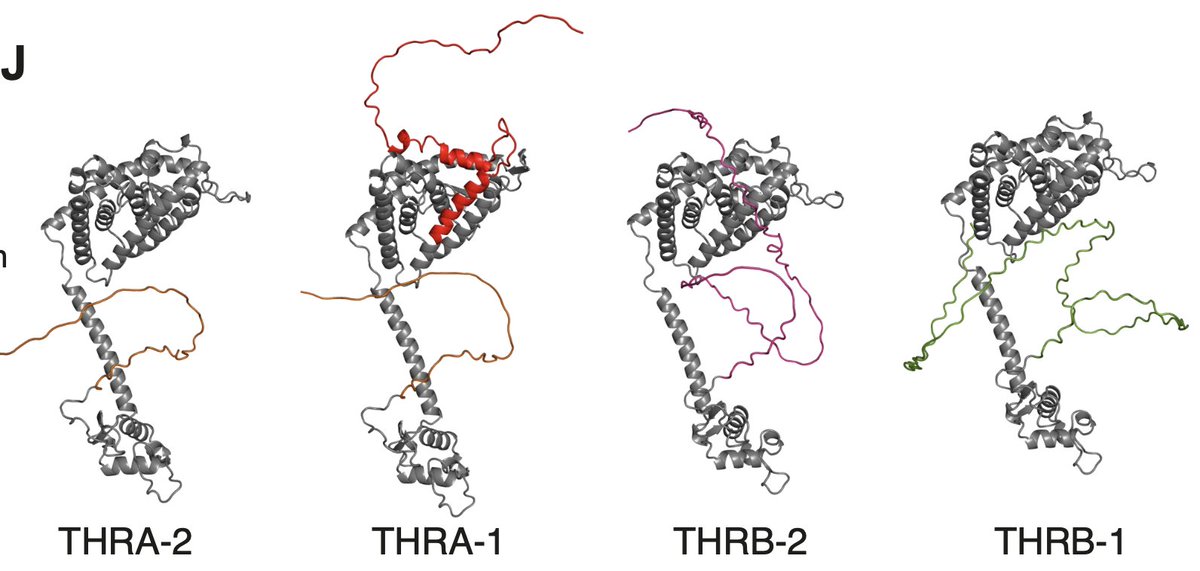
excited to share the first story from my postdoc today!! we profiled hundreds of transcription factor (TF) isoforms & found that two-thirds of alternative TF isos differ from their cognate reference iso in at least 1 key molecular activity
https://t.co/VvMjAoMhef
Based on these widespread differences in molecular functions, alternative isoforms of TFs form a crucial, but neglected, layer in the regulation of gene expression. Improving knowledge of this layer will be key to future progress in understanding cellular function and dysfunction
Of all protein-partners, it was the obligate dimerizing of TFs that was most preserved across different isoforms (e.g. bHLH-bHLH, bZIP-bZIP etc.) suggesting a need to preserve those for these alternative isoforms to function
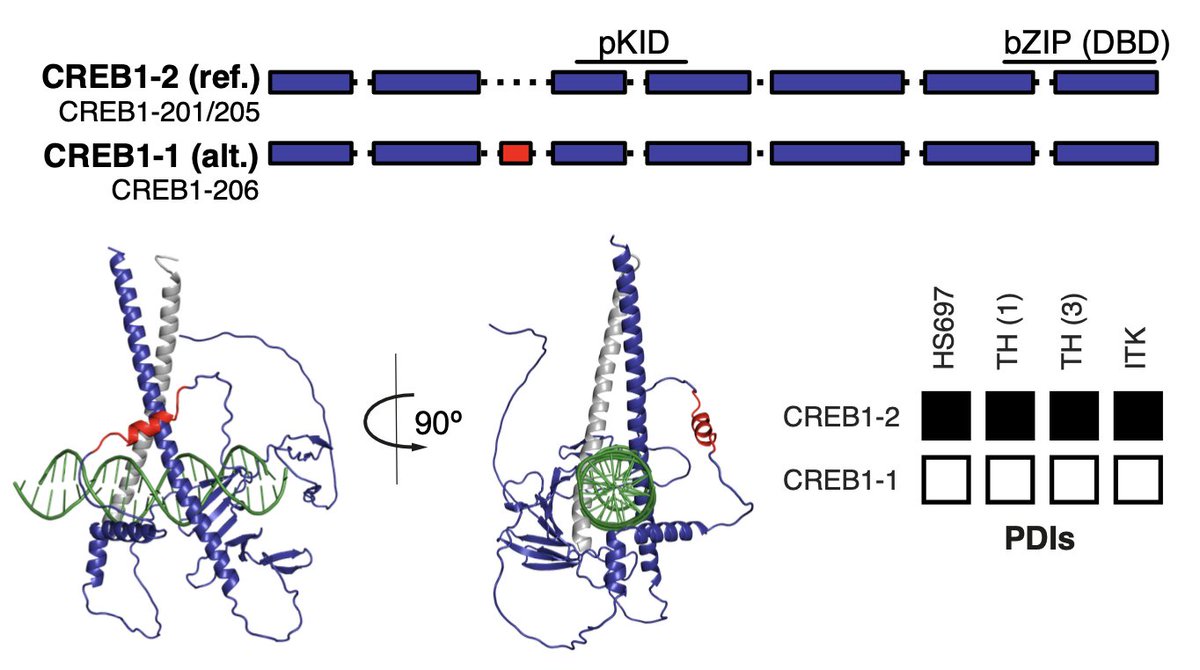
We found most alternative isoforms differed from the reference isoform of the same gene. Those differences are really challenging to predict from sequence features, e.g. differences in disordered regions far from the DNA-binding domain frequently affected DNA-binding