If you’re in the Boston/Cambridge area on Friday, December 5 (right after the Materials Research Society Fall Meeting), join us at Harvard University for our NequIP Tutorial.
👉 Please RSVP here to attend and receive event updates:
https://t.co/iRSKdtvhNt
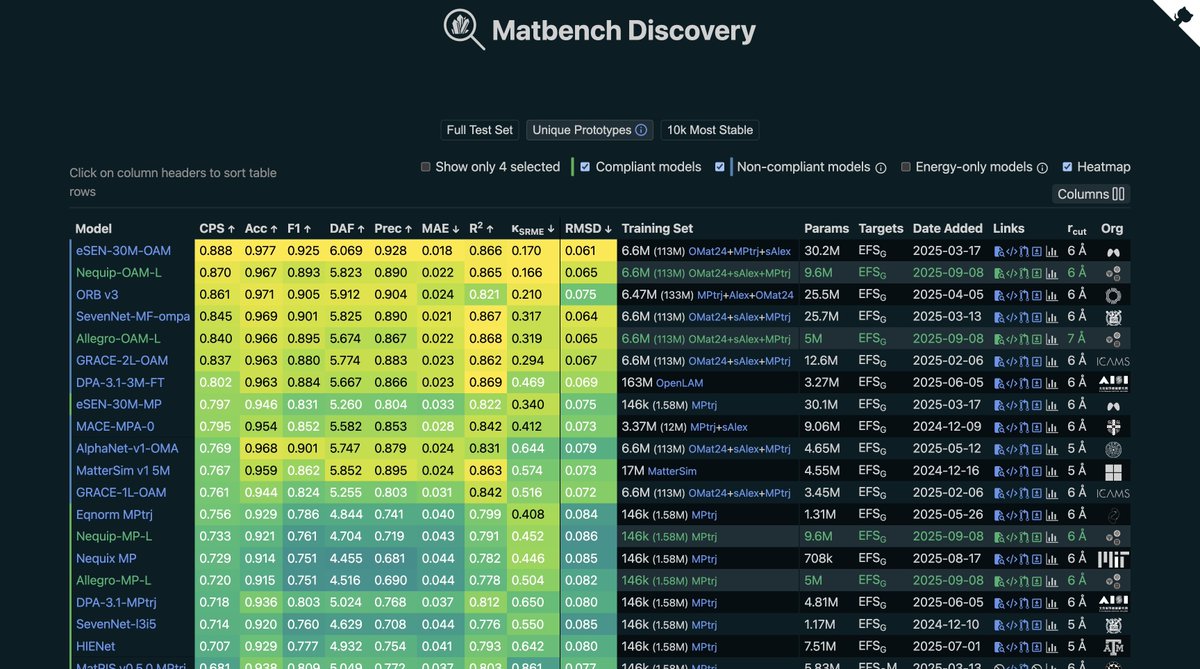
Excited to share that our NequIP and Allegro foundation potentials, trained by @Kavanagh_Sean_, are up on Matbench Discovery. Check them out at https://t.co/oLrHUBuQxm ! 🚀
Great to see our initial set of NequIP & Allegro foundation potentials released and on matbench-discovery!
Along w/excellent accuracies, we also find our model to (𝘤𝘶𝘳𝘳𝘦𝘯𝘵𝘭𝘺) be the fastest of leading foundation potentials – see our posters below 🏎️
Preprint incoming!
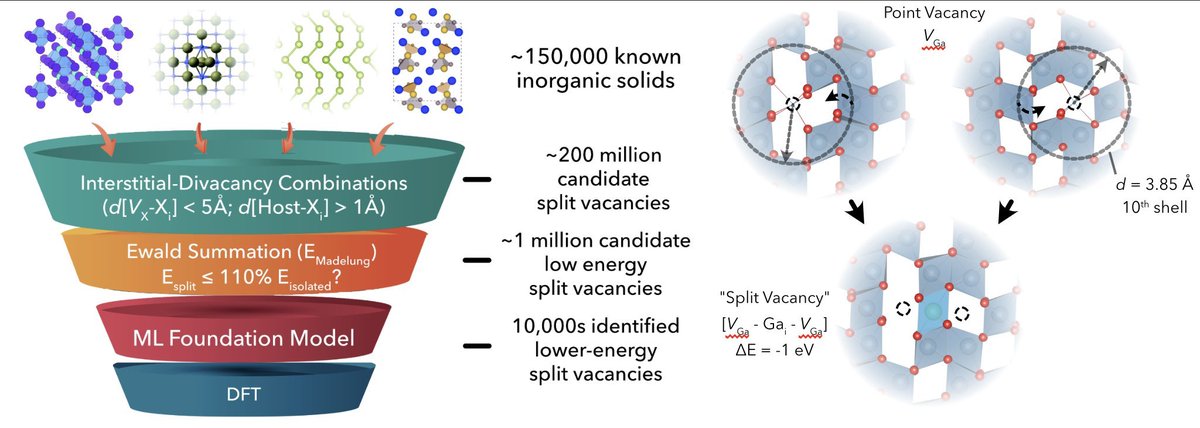
Machine learning can be powerful for understanding defects, but currently sufficient only in select cases.
MLIPs (& geometric/electrostatic tools in doped) allow screening for challenging 'non-local' defect reconstructions (split vacancies) in all ICSD/MP solids, w/caveats 🔗
@_MitKotak@AtomArchitects We look forward to adding more features. For example, we just released support for OpenEquivariance in NequIP: https://t.co/ZY5YbtGk4T.
(Thanks Vivek and Austin https://t.co/OtovKhmlyi)
Feel free to reach out if you have questions or ideas: https://t.co/68eGUoFuwR
Last month, we released a major update to the NequIP framework that fully leverages PyTorch 2.0 compilation for MLIPs. It’s significantly faster, easier to use, and more versatile than before.
Preprint: https://t.co/YuX5cOAcl7
Code: https://t.co/qfNPqWR5nU
https://t.co/TW6oJiKa5L
With @_MitKotak from @AtomArchitects, we also added custom GPU kernels for the Allegro tensor product. These combined improvements made Allegro 5-18x faster than before, and for large models, enabled simulations with 40-50 times more atoms than what was previously possible.
A machine learning framework that can predict with quantum-level accuracy how materials respond to electric fields, up to the scale of a million atoms.
https://t.co/facOvEOJZU
Allegro-Pol achieves excellent strong and weak scaling performance, enabling simulations of dielectric and ferroelectric properties of materials at the million-atom scale!
Our Allegro-Pol model extended the Allegro architecture to predict how materials respond to external electric fields while enforcing physical rules. It could describe vibrational, dielectric, and ferroelectric behavior for systems up to millions of atoms!
https://t.co/T5GImDzVak
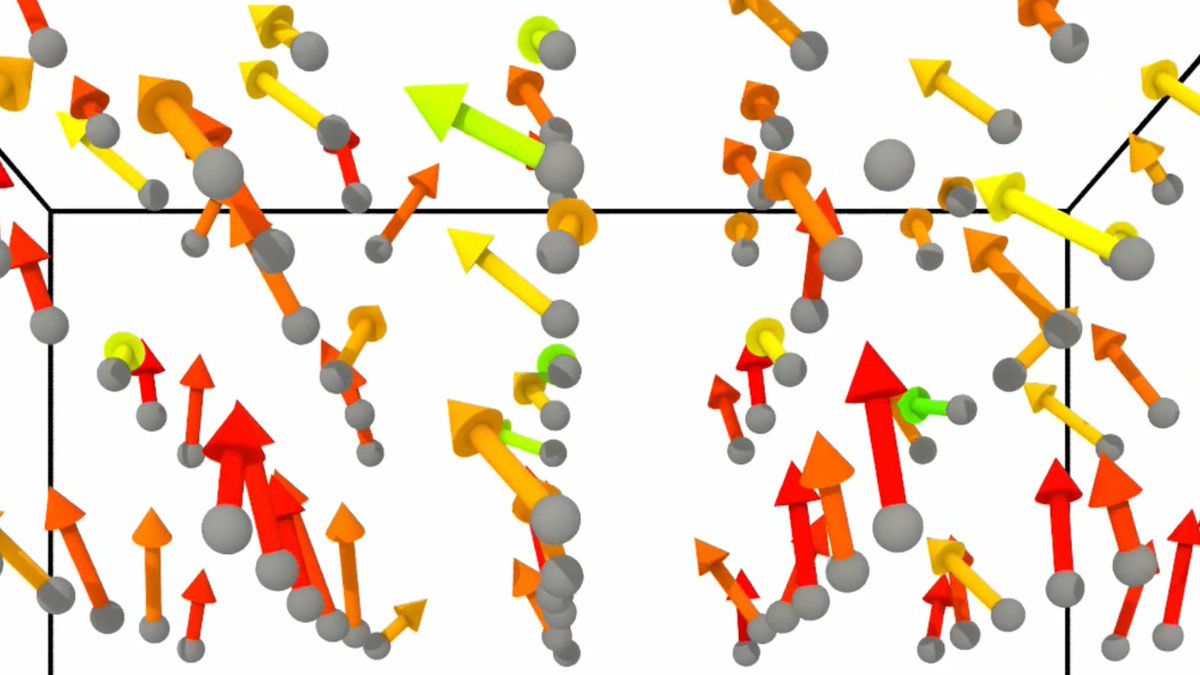
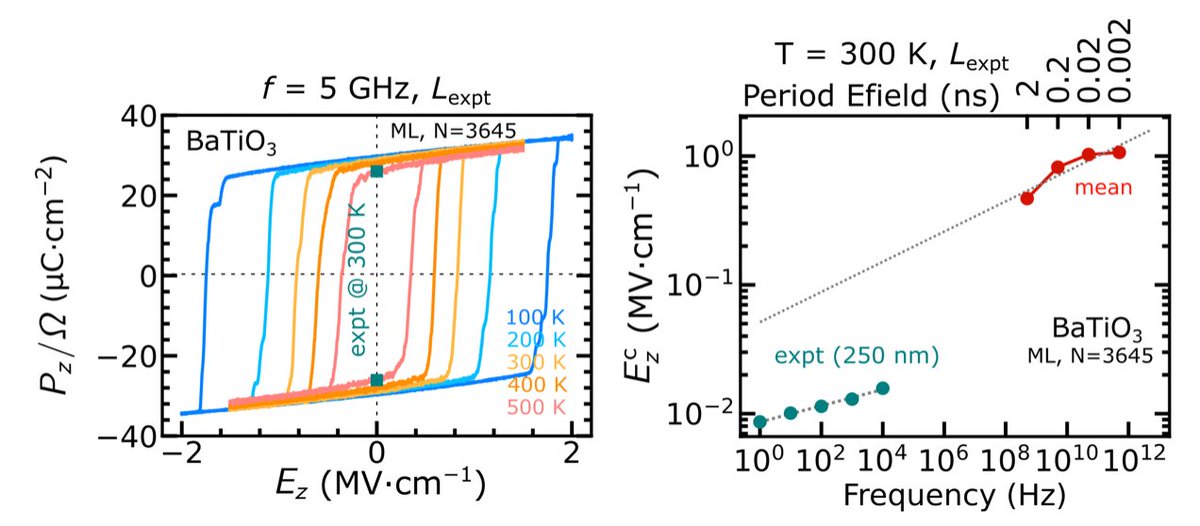
We applied Allegro-Pol to study the temperature-dependent and frequency-dependent ferroelectric response of BaTiO3, revealing the underlying mechanisms of nucleation and growth that govern ferroelectric domain switching.
Beyond happy to announce today Allegro-pol, a machine-learning framework that predicts how materials respond to electric fields with quantum-level accuracy, capturing vibrational, dielectric, and ferroelectric behavior at the million-atom scale! 🚀
https://t.co/rEKPH58Snd
Discover our simple guidelines for training accurate and transferable equivariant ML interatomic potentials for ionic liquid mixtures. Test them on your systems and let us know your results! @JPhysChem#IonicLiquids#MachineLearning DOI: https://t.co/yaPe6jLty8
💡Open position for Professor in Applied Mathematics at Harvard @hseas with focus on Computing and AI for Science, Engineering, and Society. Emphasis is on development of applications with strong mathematical and computing foundations. Apply by 12/31/23. https://t.co/Wgczp7kHNC
Beyond happy and very honored to receive my tenure promotion at Harvard @hseas. Most grateful to all my colleagues and collaborators, especially the amazing members of our @Materials_Intel group, whose work made this possible. Now the fun begins😀