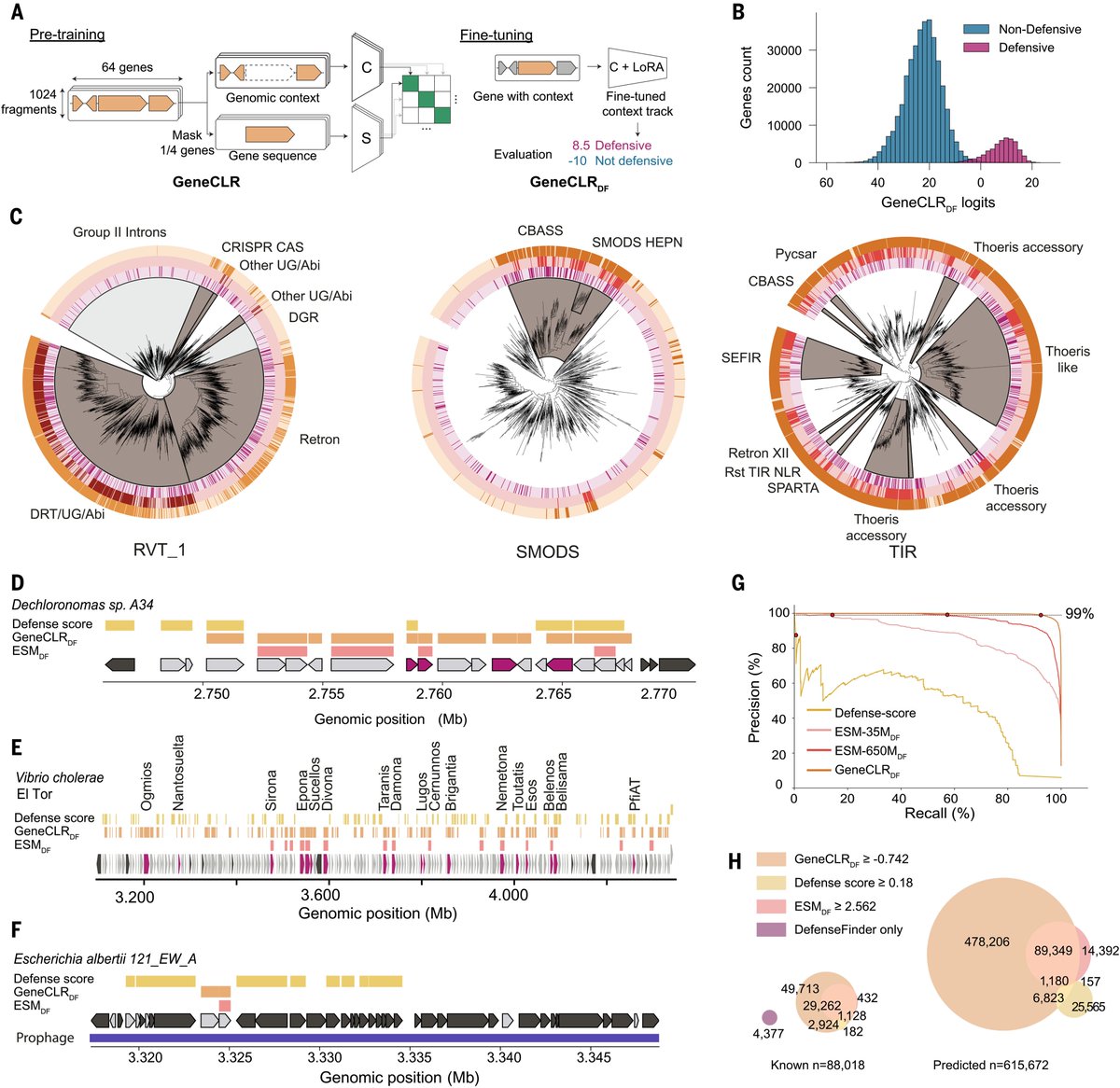
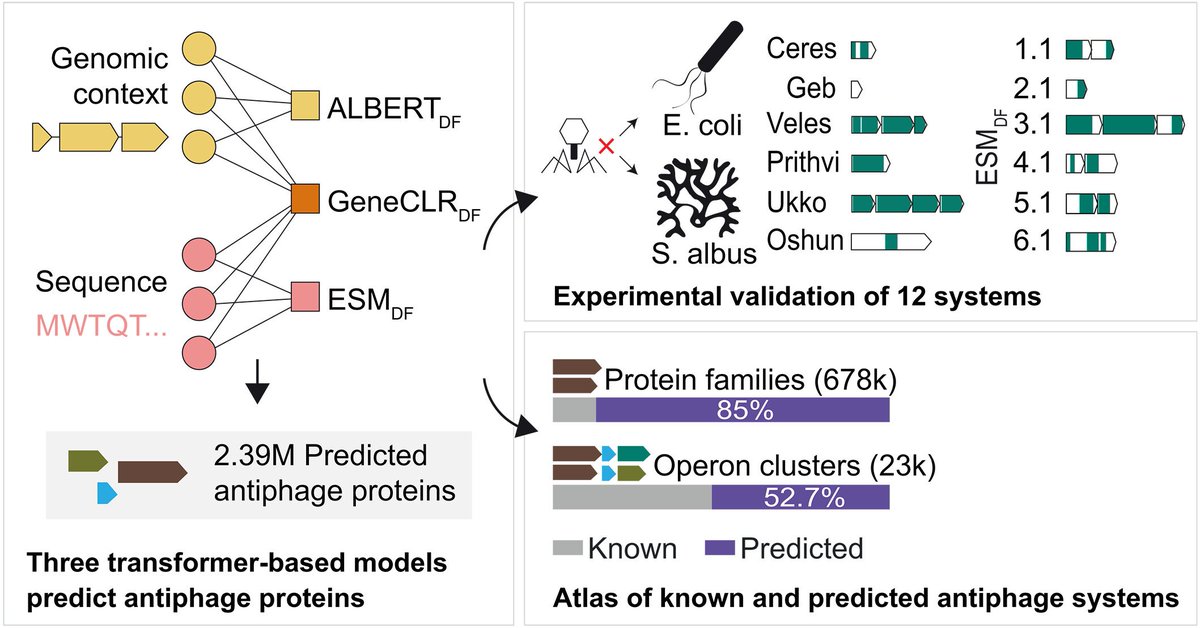
I have also included details of the benchmarking experiments. Kudos to the ESM team for their remarkable open-source work.
Executable files and source codes:
https://t.co/YonOBjakQI
Introducing a weekend project: Folding Everywhere. a pure-Rust, dependency-free re-implementation of ESMFold1/2 that predicts protein structure on the CPU with one executable, no Python, no PyTorch, no CUDA, and nothing to install!
With the help of Opus, I did a from-scratch port of ESMFold, which gives us a single native executable for Windows / macOS / Linux.
- The application itself is a single ~2 MB executable.
- On first use it automatically downloads the ESMFold weights (~8.4 GB for v1 and ~30GB for v2). No internet required afterwards.
- validated layer-by-layer against the official PyTorch model (fp32). In most cases, the error is ~0.0001 Å RMSD (fp32 round-off).
@BrianHie after reading your methods section: eGFP expression is end-point measured after 8 hours after “normalizing to OD of 1”. Any OD over 0.5 is early stationary phase. Cells slow down growth. eGFP expression saturates (repressed or not). Lower fold-change in expression.
@natolambert@Ishaank1999 This is as bad as it gets! Every day of delay in finding cures costs many lives, totally on Anthropic! They are antagonistic to humanity not just China!
We are releasing Carbon: a crazy fast DNA model
Carbon is 275x faster than the next best model. So fast you can process the whole human genome on a single GPU in <2 days.
Here are the tricks we used:
When modelling DNA sequences a lot of the performance comes down to tokenizing the sequences in a smart way. BPE tokenizer struggle because there are no whitespaces and character (called base in DNA) level tokenizers waste a lot of compute on too many tokens.
Carbon is built with a unique tokenizer: we split sequences in chunks of 6 bases, but during both training and inference we can work with single base resolution. That's similar to having word tokens but resolving them at the character level. All possible thanks to the DNA tokens unique structure.
The architecture combined with the tokenizer makes the model 275x faster than the previous SoTA (Evo2) at this size.
We built an interactive demo so you can explore how the model can generate DNA sequences, investigate the structure of genes, predict the effect of mutations, generate and fold proteins and even reconstruct parts of the tree of life.
https://t.co/OWEUoxAFjG
EVO2 has been used for some exciting applications like viral genome design & discovery of new phage defense systems. But in both cases, I'm pretty sure models trained only on restricted clades would do just as well or better. 6/
Our preprint for de novo DNA binder design is out! https://t.co/fWBls0vEzQ. The punchline: methods have gotten good enough that we can find sequence specific DNA binding proteins from screening as few as 96 designs per target.
Why do cloning tools still suck? This problem seems like a low-hanging fruit for AI to solve.
Today, if a scientist wants to make a new plasmid or DNA sequence, they often go into their freezer, figure out which DNA sequences they have, upload those DNA sequences to Benchling (or another platform), and then must figure out how to "convert" those sequences into what they want. Should I do Golden Gate or Gibson Assembly? What annealing temperature should I use for my primers? And so on.
There are already tools that help with each of these steps, but has anybody "automated" this decision-making? If so, I'm not familiar with them. (A tool called J5 is probably the closest thing, but it won't recommend the optimal method given a scientist's existing sequences and primers.) And if the scientist makes even one error in this multi-step design process — like forgetting about an internal restriction site in a gene — they basically waste an entire week of work.
(You might object to this and say, “But DNA synthesis solves this problem; just synthesize the full plasmid directly!” But people have been saying that for decades at this point, and DNA synthesis costs have not fallen in several years. Cloning DNA remains essential.)
What we need is a fully automated, end-to-end cloning design tool that selects the best method based on a library of existing sequences and primers; a tool that recommends the optimal approach based on cost, speed, and so on. “Design tools” for cloning may not seem like a sexy thing to work on, but whoever solves it will marginally improve the lives of many scientists.
With this in mind, I’ve given $1,500 in microgrants, courtesy of Astera Institute, to two people — Jai Padmakumar and Xavier Bower — who have been thinking about this problem. Bower has already built an open-source prototype, called IceCreamClone. (Visit icecreamclone[dot]xavbio[dot]com to see a demo.)
Here's how a tool like this should work:
First, you specify the plasmid you want to build. Then, you upload your current plasmid library, a collection of DNA sequences already in your inventory, and existing primers. The tool takes these data and outputs multiple cloning protocols based on different metrics, such as lowest cost, fastest speed, or the protocol most likely to be successful. The tool also runs a series of checks on all the sequences to make sure they don’t have internal restriction sites, for example, or weird secondary structures.
It would be particularly cool if scientists using this tool could opt-in to sharing their data. The tool could then prompt them afterwards: How did the cloning go? Can you upload the results? Over time, this feedback data could be used to train predictive models that make cloning far more likely to be successful.
Of course, there are issues with this idea. For one, it requires that people upload their entire catalog of existing sequences + primers, which is quite tedious for some laboratories; especially those with decades of cloning experience. Ideally, these tools would directly integrate with Benchling and Addgene.
Anyway, I continue to think this is a "low-hanging" problem worth working on. Whoever makes an easy-to-use, end-to-end cloning design tool with really good predictive accuracy could presumably make a small business out of it. And, in doing so, you'd make many people happy!
🥳 Huge preprint 🔔 Today we share something our group has been working toward for a long time, led by
@LucasMoriniere We asked can we predict which receptor a phage targets from its genome sequence alone? For most phages, we couldn’t. So Lucas set out to do something crazy!
I made a list of a bunch of organisms and I will be reading each of them out loud in Latin laticus. The goal is to say at least one nice thing about each of them.
Join me, Josh Jones-Dilworth, and Elizabeth Bennett from Revive and Restore at @sxsw for our session on Curious Critters & Human Health.
A non-hyped explainer of the “cell simulation” paper.
The recent study about the “4D” simulation of a minimal cell has been getting a lot of attention on social media. Unfortunately, most posts about it have serious errors. I’ve seen people claim that the model simulates every chemical reaction in the cell, for example, which is not true.
Some biomolecules and reactions *are* tracked individually in the simulation, including proteins and RNA (and ribosomes), and the chromosome. But the simulation does not track individual metabolites (like ATP or glucose), water, nucleotide precursors, lipds, and so on. These "other" molecules are represented, instead, as concentrations (using ordinary differential equations).
But anyway, here goes my quick explanation:
Researchers built a computational model that simulates roughly 100 minutes of biological time, or one cell division, for a single bacterial cell. Each simulation takes 4–6 days to run on two NVIDIA A100 GPUs, and the authors ran it 50 times in replicate. The cell simulation includes some elements of randomness, so each replication attempt leads to a slightly different outcome. When they plotted out these replicates and averaged results, they found that the model could predict a few things without being fitted to experimental data: The simulated cells “divided” every 105 minutes, on average, which matches experimental results; and the mRNA molecules had an average half-life of 3.63 minutes, which is roughly what we’d expect from experiments, too.
The cell they are modeling is called JCVI-syn3A, and it is not a naturally-occurring organism. It’s a bacterium that has been engineered, over many years, to have a small genome. It only has 493 genes (compared to 4,000+ for E. coli), all of which are housed on a single chromosome. The Syn3A cell was made by taking a natural organism, called Mycoplasma mycoides, and then slashing out non-essential genes. Its entire proteome, transcriptome, and metabolism have been studied in depth, which is why it’s being used to build these whole-cell simulations.
The actual *simulation*, though, is not a single thing! Instead, the authors wrote down all the “stuff” that happens inside a cell (transcription! translation! metabolism! lipid biosynthesis!) and decided which type of mathematical model would be best-suited to describe each thing. Some cell processes were modelled deterministically, others had “spatial” elements, and other parts were relatively random.
More specifically, they used four different types of models to build this simulation:
1. A Reaction-Diffusion Master Equation, which was used to model the individual proteins, RNAs, and ribosomes.
2. A Chemical Master Equation, which was used to model things where spatial location doesn’t matter as much (it basically treats the whole cell as one mixed entity); including tRNA charging.
3. Ordinary Differential Equations, which you may be familiar with from Calculus class, were used to model changes in ATP concentration, lipids, and so on.
4. Brownian Dynamics, which simulated the chromosome as a physical chain of beads, where each bead represents 10 base pairs of DNA.
The Reaction-Diffusion Equation works like this: Basically, they chopped up the entire digital cell into a 3D grid of cubes. Each cube measures 10 nanometers on each side. The whole cell is about 500 nanometers across, so there are tens of thousands of cubes in the cell's interior. (This is a useful way to coarse grain the simulation; if the cubes were smaller, the simulation would take much longer to run.)
Each cube is a little box that contains some number of molecules. At every “step” in the simulation, only one of two things can happen to the molecules in each box: Either they react with a molecule in the same box, or they diffuse (“hop”) to an adjacent box. That’s it; the model is just rolling a die for each molecule at each time step in each box, and using those results to decide how each molecule changes over time.
(The reason this spatial model is important is because biology only works if molecules physically bump into each other. And so this spatial grid means that, unlike simpler models, a protein actually has to “diffuse” across boxes in the cell to encounter its reaction partner; only then can it react and do something useful.)
So anyway, each of these models is used to represent a different type of molecule. It’s not like there is a single, all-powerful simulation that they are running here; instead, they’re running these four models together, using a script that synchronizes their results with each other.
The Reaction-Diffusion equation is the main part of the simulation. It takes time steps of 50 microseconds of biological time. Every 12.5 milliseconds of biological time — meaning every 250 RDME steps — the simulation pauses so that the other models can synchronize based on the latest state of the simulation. The Brownian Dynamics part runs on a completely separate GPU, and only updates every four seconds of biological time.
So that's the gist here. But let's also be honest about what this simulation does NOT do:
- It does not include polysomes, which are a cluster of ribosomes that all latch onto a single mRNA and translate at the same time. Polysomes are really common inside of cells, but this simulation assumes that each mRNA can only be translated by one ribosome at a time.
- It does not include polycistronic transcription. In bacteria, genes are often grouped next to each other on the chromosome and thus “transcribed” (or turned into mRNA) all at once, together. The majority of genes in E. coli, for example, are arranged in these operons, and the authors of this paper acknowledge that many Syn3A genes are likely co-transcribed the same way. But the simulation doesn't capture it.
- The authors manually tuned many parameters to get the model to make predictions that more closely resemble experiments. Earlier simulations were waaaayyyyy off from experimental results. For example, they adjusted the ratio of mRNA binding rates to ribosomes versus degradosomes because, in earlier simulations, mRNA was being degraded too quickly, before ribosomes could translate it, causing most proteins to be severely underproduced.
- In the Brownian Dynamics model, the authors added a “fake” 12 pN physical force to push the two daughter chromosomes apart during division, because the real biological mechanism for chromosome partitioning in Syn3A is not known.
- And some other things.
That being said: This model is really cool! I love papers like this! I'm enamored by scientists who choose really difficult problems (like simulating an entire cell) and actually go after it and make progress!
This paper is amazing because it shows us what we are able to simulate well, and what we don't yet understand, and to figure out which experiments we ought to perform to reconcile the two. So instead of framing this paper as "OH MY GOSH SCIENTISTS FIGURED OUT HOW TO SIMULATE AN ENTIRE CELL!" we should frame it as proof that there is still plenty of room at the bottom, many measurements to be made, and many avenues to explore as we seek to understand biology better.
Can we simulate realistic evolutionary trajectories and “replay the tape of life”? In this work, we propose a flexible, generalizable framework for modeling how the entire protein seq evolves over time while capturing complex interactions across sites. 1/n
https://t.co/DIwQ40C478
Excited to announce our latest work! We present ARSENAL, a short-context DNA language model specifically designed to learn important sequence features in noncoding regulatory DNA.
Why is such a model important? Read on to find out!
https://t.co/3FyTSxxe5K
Plant and soil microbe engineering is a rapidly growing field. This "review" was written as a comprehensive guide to their genetic engineering, from the idiosyncrasies of promoter design to whole genome synthesis.
https://t.co/bm2wZhcCfv
So you sequenced a billion+ bases. That don’t impress me much.
Over the last decade, biologists have learned how to generate genome sequences at extraordinary scale. What hasn’t kept pace is figuring out what those genes actually do.
Today we infer what a gene by computing similarities, whether by sequence or by structure. Incredibly useful for a first pass but not good enough in most cases to understand or engineer biology. You have to modify the genes and measure the consequences to know what they do.
JERBOA is a toolkit to figure out what genes do at scale. We've used it to unlock 43 non-model microbes across 12 different phyla.
Molecular assets on @Addgene. Analysis software on @github. There's so much to explore, get out there!!























![NikoMcCarty's tweet photo. Why do cloning tools still suck? This problem seems like a low-hanging fruit for AI to solve.
Today, if a scientist wants to make a new plasmid or DNA sequence, they often go into their freezer, figure out which DNA sequences they have, upload those DNA sequences to Benchling (or another platform), and then must figure out how to "convert" those sequences into what they want. Should I do Golden Gate or Gibson Assembly? What annealing temperature should I use for my primers? And so on.
There are already tools that help with each of these steps, but has anybody "automated" this decision-making? If so, I'm not familiar with them. (A tool called J5 is probably the closest thing, but it won't recommend the optimal method given a scientist's existing sequences and primers.) And if the scientist makes even one error in this multi-step design process — like forgetting about an internal restriction site in a gene — they basically waste an entire week of work.
(You might object to this and say, “But DNA synthesis solves this problem; just synthesize the full plasmid directly!” But people have been saying that for decades at this point, and DNA synthesis costs have not fallen in several years. Cloning DNA remains essential.)
What we need is a fully automated, end-to-end cloning design tool that selects the best method based on a library of existing sequences and primers; a tool that recommends the optimal approach based on cost, speed, and so on. “Design tools” for cloning may not seem like a sexy thing to work on, but whoever solves it will marginally improve the lives of many scientists.
With this in mind, I’ve given $1,500 in microgrants, courtesy of Astera Institute, to two people — Jai Padmakumar and Xavier Bower — who have been thinking about this problem. Bower has already built an open-source prototype, called IceCreamClone. (Visit icecreamclone[dot]xavbio[dot]com to see a demo.)
Here's how a tool like this should work:
First, you specify the plasmid you want to build. Then, you upload your current plasmid library, a collection of DNA sequences already in your inventory, and existing primers. The tool takes these data and outputs multiple cloning protocols based on different metrics, such as lowest cost, fastest speed, or the protocol most likely to be successful. The tool also runs a series of checks on all the sequences to make sure they don’t have internal restriction sites, for example, or weird secondary structures.
It would be particularly cool if scientists using this tool could opt-in to sharing their data. The tool could then prompt them afterwards: How did the cloning go? Can you upload the results? Over time, this feedback data could be used to train predictive models that make cloning far more likely to be successful.
Of course, there are issues with this idea. For one, it requires that people upload their entire catalog of existing sequences + primers, which is quite tedious for some laboratories; especially those with decades of cloning experience. Ideally, these tools would directly integrate with Benchling and Addgene.
Anyway, I continue to think this is a "low-hanging" problem worth working on. Whoever makes an easy-to-use, end-to-end cloning design tool with really good predictive accuracy could presumably make a small business out of it. And, in doing so, you'd make many people happy!](https://pbs.twimg.com/media/HF9WynObYAAc376.png)