@Miguel_RuizC@drjosephpowell Hi @Miguel_RuizC it's hard to define a 'normal' range as different cell types vary in their nuclear fraction values. I'd recommend comparing NF values within all cells of a given type to identify cell-free droplets/damaged cells
Australian women took a good look at the prime ministerial bulldozer... and sent him a John Deere letter. Here's my column on why https://t.co/IjkqjaDpiK
Many intercellular communication systems (BMP, Notch, Wnt, FGF, etc) use sets of promiscuously interacting ligand and receptor variants rather than seemingly simpler one-to-one architectures. Why? Our 2 papers grappling w/this question just out in @CellSystemsCP
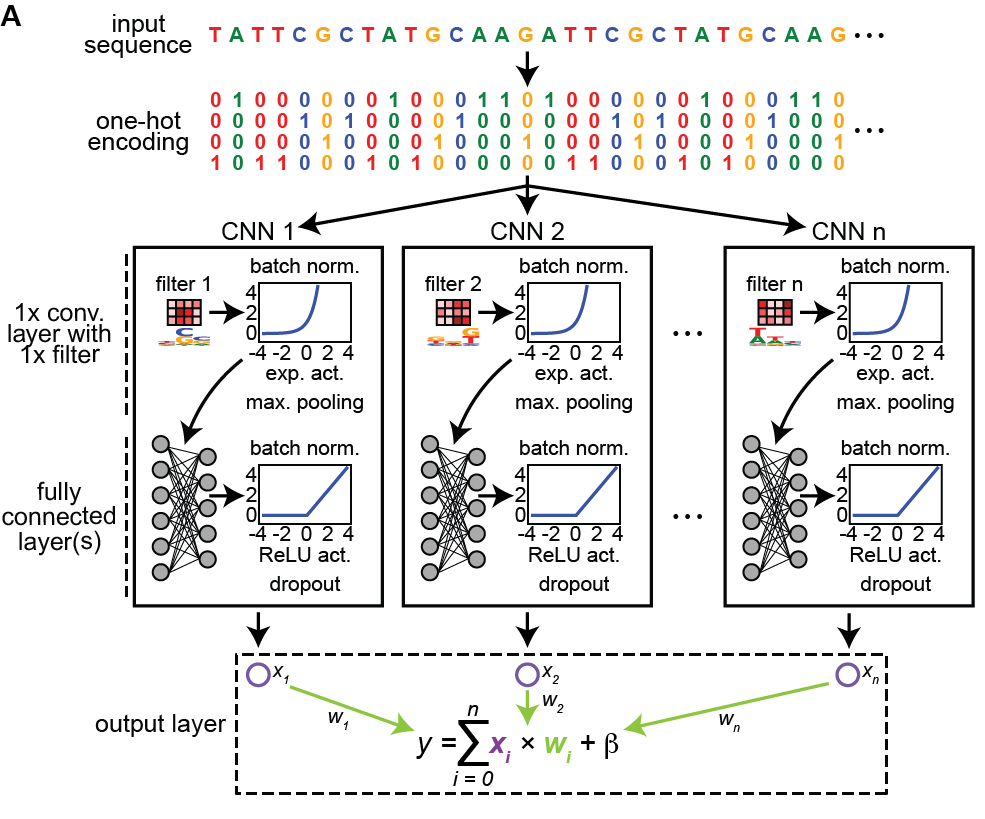
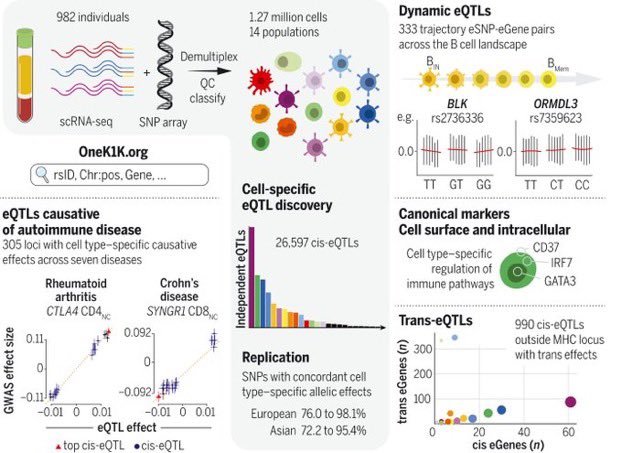
Our work “Single-cell eQTL mapping identifies cell type-specific genetic control of autoimmune disease” just published in @ScienceMagazine. The work started with the creation of the OneK1K cohort, consisting of scRNA-seq data on PBMCs from ~1000 donors https://t.co/Uzskay7TeM 🧵
@ChiragNepal@tangming2005 I don't think MALAT1/NEAT1 enrichment is a consequence of cell damage. I think the pool of RNA becomes depleted of cytoplasmic transcripts lost through the damaged cell membrane, so the nuclear-retained transcripts will make up more of the pool of sequenced RNA from those cells
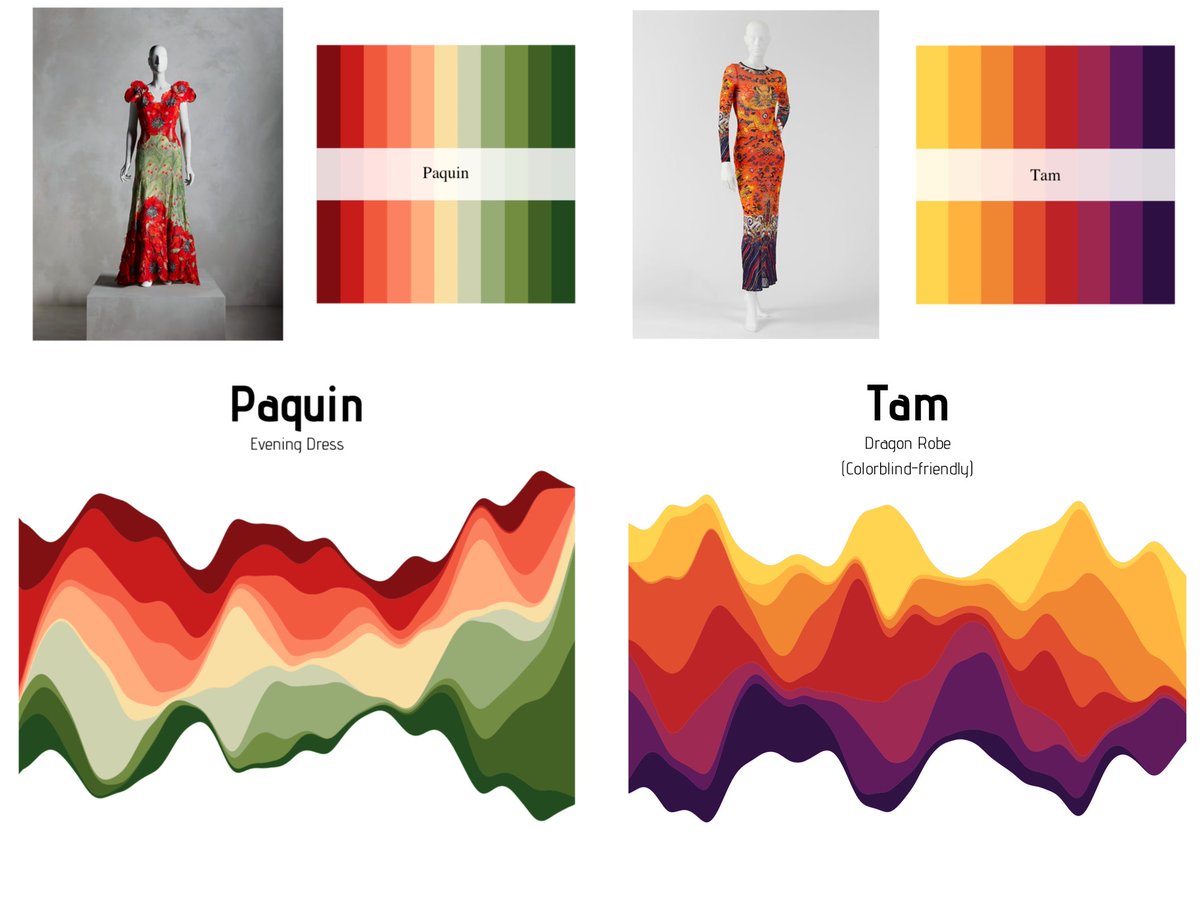
✨✨ PACKAGE UPDATE ✨✨
MetBrewer 0.2.0 is officially out 🥳 Available both on Cran and through my GitHub here: https://t.co/wYHMEKoJLH
New Palettes are out and some amazing new features. Full description of what has changed below :)
#MetBrewer#rstats#r4ds#dataviz
Have you ever wondered which demultiplexing and doublet detection methods to apply to your single-0cell dataset? Wait no more! We've developed Demuxafy, an easy-to-use software platform to support method selection and execution for single-cell demultiplexing and doublet removal.
Introducing Demuxafy: Improvement in droplet assignment by integrating multiple single-cell demultiplexing and doublet detection methods. Work led by @drneavin as part of the sceQTL-gen consortium https://t.co/Dr8qOmjpWF
1/ single-cell RNAseq data matrix is sparse. dominant 0s makes gene-gene correlation calculation hard. Tools that I know to tackle this problem #scRNAseq :
https://t.co/6LfiwAIXUU
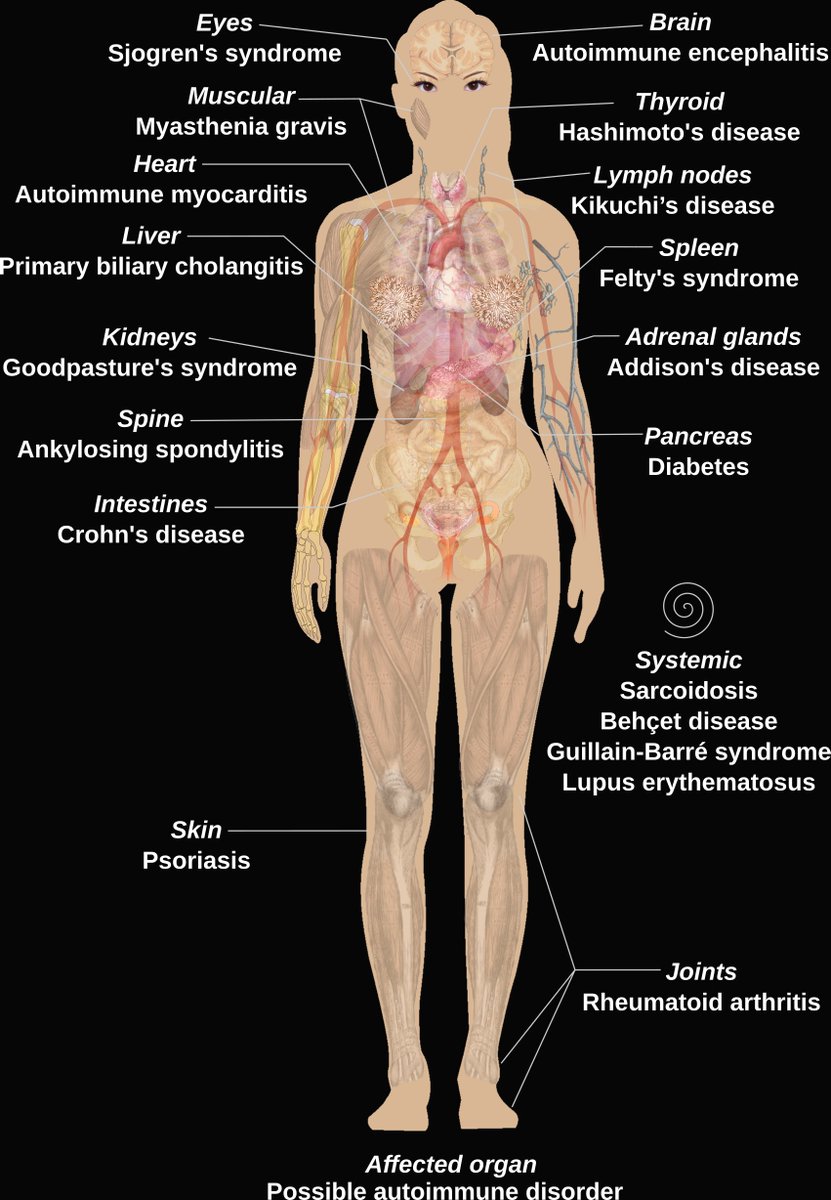
Milestone alert 🚨 the #ChronicIllness#PatientAdvocacy project has hit 1000 participants!
Thank you all for chronicling 162 illnesses, snapshot below where text size = number of responses.
Sneak peek of what your responses are showing us, a 🧵… 1/
#InvisibleIllness#NEISvoid
Interested in lncRNAs? Check out our new paper up on bioRxiv where @MK_CCI and I use high temporal resolution RNA-seq time series data to demonstrate both human and mouse lncRNAs mirror the expression of nearby protein-coding genes https://t.co/Od8EYrtXlN