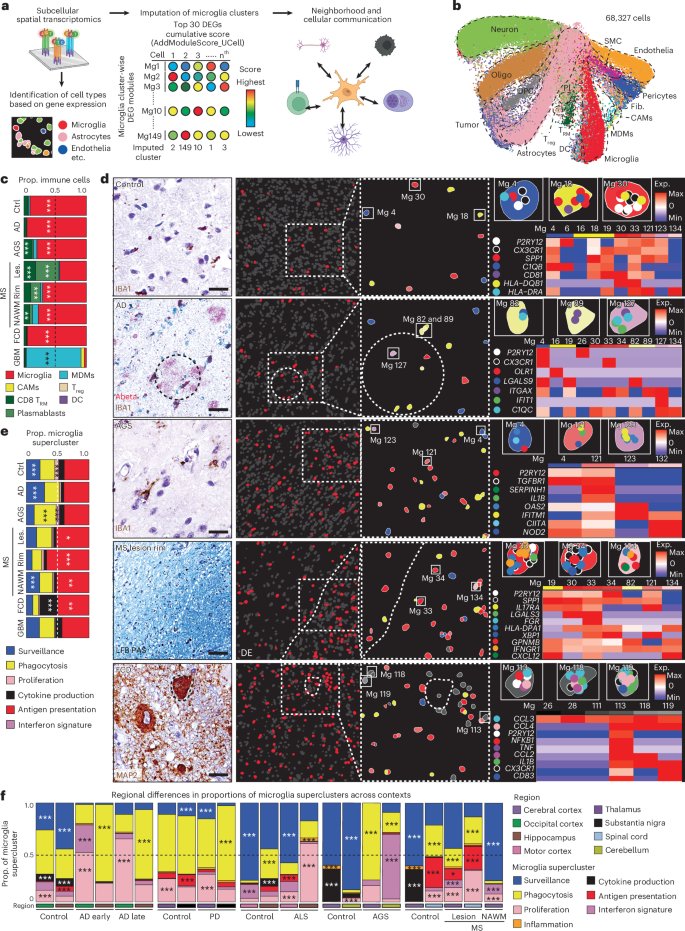
Grateful to see this one out! Thank you to our collaborators at Eisai and to @ErikMusiek for his mentorship and leadership throughout this project. @WashUNeurology
Very excited that @asharma313's paper on orexin antagonist lemborexant, amyloid pathology, and microglia is out! A great collaboration with scientists from Eisai with clear translation relevance for AD prevention. @WashUNeurology@WashUcobras
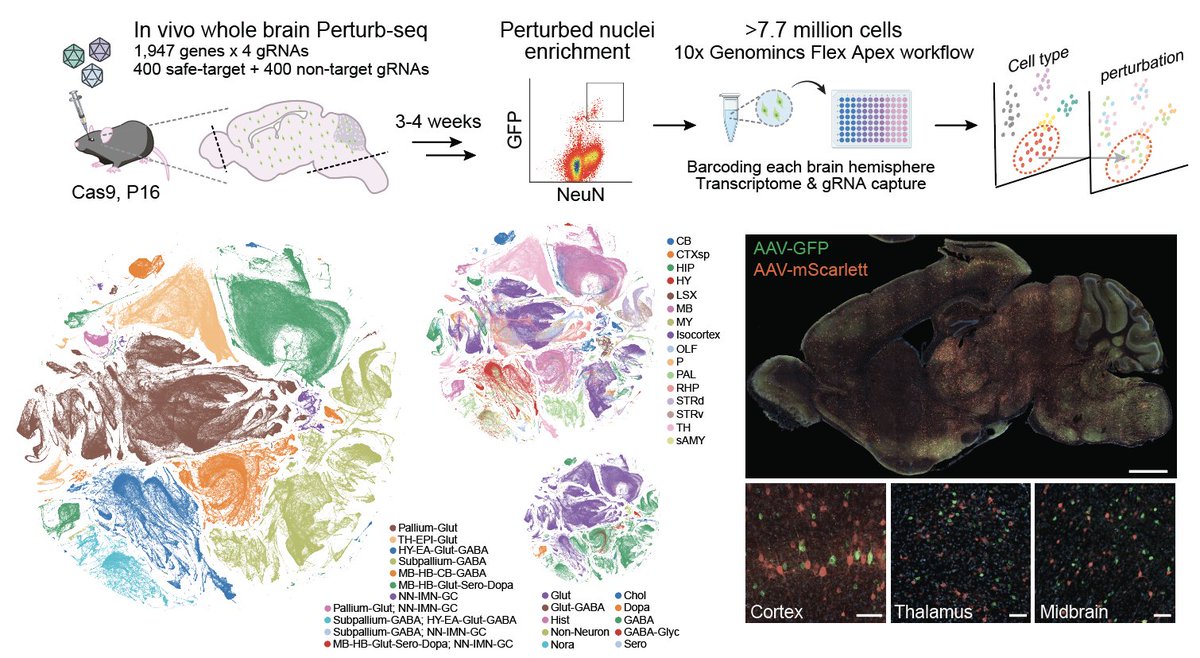
📢 Preprint: we present a whole-mouse-brain in vivo Perturb-seq atlas, 7.7 million cells, 1947 disease-associated perturbations, moving toward direct readout of how human genetics rewires cell states & circuits in vivo. Grateful for the Team! @NVIDIAHealth https://t.co/01c1KFuLFw
AI is cool and all... but a new paper in @ScienceMagazine kind of figured out the origin of life?
The paper reports the discovery of a simple 45-nucleotide RNA molecule that can perfectly copy itself.
Genetics of human longevity more important than was previously thought! Heritability of intrinsic human life span is about 50% when confounding factors are addressed | Science https://t.co/MW4VhJuiJQ
“Show me the mechanism of action.”
“Uh. from the p-p-perturb seq?
right here. Knocks down Gene X, and the cells shift into Cluster 7."
“You used fkn UMAP again. FUCK. Zoom in.”
“Sorry?”
“Zoom. The fuck. In.”
“…Okay.”
“Do you see it?”
“…See what?”
“He doesn’t see it. The cell biology postdoc from Stanford does not know how to see a cell. He stares at a two-million-cell embedding and doesn’t see the conflations he just planted in my S3 bucket.
WHERE is the temporal resolution?”
“The… what?”
“Where is the time? You hit the cell with a perturbation and you measured it once. Once. That’s not a mechanism. That’s a fucking POSTCARD!”
“We sequenced at 72 hours. That’s standard.”
“Standard is not the same as correct. You are aliasing causality. You compressed a dynamic process into a static endpoint and you’re think you will cure metastatic cancer.”
“But the differential expression is significant.”
“WOW! DIFFEWENTIAL EXPRESSION IS SIGNIFICANT! WOW! WHEN THE FUCK IS IT NOT.
Yes, because statistics is Mario Kart.
A fantasy land where causality is optional and variance disappears if you collect enough cells. Real biology has inertia.
Feedback.
Competing pathways.
Do you understand the difference between correlation and mechanism? Or did you flunk out of STAT 101?”
“…I mean, we saw Gene X regulate Pathway Y.”
“No. You saw Pathway Y exist in the same cell after you kicked it down the stairs and waited three days. That’s not even a crime scene, you dimwit. That is a post-mortem autopsy.”
“The model inferred a trajectory--”
“STOP the buffoonery. Do not blame the model.
The model is a mirror.
In this particular case, you can see it mirrors the clusterfuck you just created in my biosafety cabinet.
If the reflection is warped, it’s because your measurement is warped.”
“Pull up the raw counts.”
“…Okay.”
“Scroll. Cell 14,982. Read it to me. What does it say?”
“Uh… mitochondrial genes up, ribosomal genes down-”
“And?”
“…And stress response markers?”
“Yes. Because you poisoned the cell and waited long enough for it to panic.
Where is the early signaling?
Where is the metabolic inflection?
Where is the first irreversible decision?”
“We don’t capture that.”
“Exactly. You built a platform that cannot see the mechanism. YOUR PLATFORM IS FUCKING BLIND.”
“The virtual cell...showed the perturbation effect cleanly.”
“Yes. because your VIRTUAL CELL IS VIRTUAL. it assumes the cell is a bag of transcripts. Do you think metabolites show up? OR AN ISOFORM? AN ISOFORM, THAT WILL DEGRADE IN THE CELL BEFORE YOU EVER REACH YOUR SENSOR? THAT ONE? YOU THINK THAT'S HOW CELLS WORK?”
“So what do you want me to do?”
“Delete the atlas.”
“What?”
“Delete it. The whole thing.”
“But that’s the core result.”
“It’s three weeks of garbage narrative built on a blind instrument. Delete it.”
“…Now what?”
“Now you rebuild the measurement. You observe the cell while the perturbation propagates. You track chemistry, not just transcripts. You capture the first divergence, not the final corpse.”
“That’s… not perturb-seq anymore.”
“Exactly.”
door slams
Our new paper by Melvin King @MKing629 is out! GABAergic Bmal1 KO reduces amyloid plaques and p-tau in 5xFAD mice- but only in LD! In DD, 5xFAD mice have fewer plaques and GABergic Bmal1 KO has no effect. Suggests light-clock interaction controls Abeta. https://t.co/x57r1rgQ5J
#PR Our Ms on brain atlas of circadian activity is out in Science! ~80%(508 regions) of whole brain show significant oscillations. We envisioned it at first CUBIC papers in 2014. It took a decade. Congrats, Katsu&Fukuaki! Ms→https://t.co/VZNUIEsHbm
DB→https://t.co/01evod4ChG
Excited to announce our collaboration with Ron Anafi at Penn is out in @Neuron! Congrats to first authors Henry Hollis & @asharma313. Our team examined circadian rhythms in human snRNAseq data from AD patients using CYCLOPS, implicating ribosomes.
https://t.co/errYRXBayn
As biomarker tests for Alzheimer’s disease are increasingly being used in clinical care, there are some key differences to note about biomarkers in clinical care and research:
1️⃣ Clinical amyloid PET scans are not the same as research amyloid PET scans. In clinical practice, amyloid PET scans are visually interpreted as positive or negative by a person and only the positive/negative result is provided in the clinical report; quantitative values are rarely provided, although some expert centers are working to add quantitative values. A recent large study showed ~86% agreement between clinical amyloid PET visual reads and quantitative values.
2️⃣ Tau PET scans are almost never performed in clinical practice in the US because they are rarely reimbursed by insurance and extremely expensive.
3️⃣ Clinical tests are primarily indicated for clinical decision making that impacts patient management (e.g., to aid with diagnosis, which may affect further testing and treatment), not to “follow” patients. For example, clinicians do not periodically perform a brain MRI or PET scans just to “follow” disease progression because we do not expect that additional scans would affect patient management.
4️⃣ In research, blood biomarker panels are analyzed using algorithms, but in clinical practice, interpreting multiple blood biomarkers is challenging due to their varying diagnostic importance. For example, p-tau217 is highly accurate in identifying amyloid pathology, while p-tau181 and Aβ42/40 offer minimal or no added value. Most clinical blood test panels (unlike PrecivityAD/PrecivityAD2, which use weighted algorithms) do not prioritize biomarkers, potentially causing confusion about amyloid status due to unclear interpretation of individual test results when results are discordant. A single, high accuracy test is often best and easiest to interpret.
5️⃣ Performing more test modalities is not necessarily better, especially if the certainty of Alzheimer’s is very high following the first test. Performing a second test in patients with a very high likelihood of Alzheimer’s may lead to a significant number of false negative results.
6️⃣ A test doesn’t have to work perfectly in everyone, as long as we know who is at risk for an inaccurate result. For example, it is very simple to identify patients with severe chronic kidney disease, which affects plasma p-tau217 levels, and choose CSF or PET for those patients.
7️⃣ Continuous values are important to clinical certainty and data on the distribution of values is helpful. This helps clinicians to know whether a value is borderline abnormal or very abnormal.
8️⃣ Diagnostic accuracy is not just based on the biomarker test sensitivity/specificity. Clinicians can integrate biomarker values with clinical information to achieve high diagnostic accuracy/positive predictive values for Alzheimer’s disease.
Amyloid-PET Quantification and Real-World Visual Reads https://t.co/CFqBIrBOcs