Excited to report the first de novo enzyme catalyzing two of the most energetically demanding reactions in biology—phosphomonoester and phosphodiester hydrolysis—with catalytic efficiencies comparable to natural enzymes! 🚀
desB was designed zero-shot with dEVA. No structure prediction, no pre-defined motif, no reaction-intermediates. 🧵
@StanfordBiosci@bioe_stanford@SLAClab@EPFL@hes_so@simonduerr
From her hospital bed, Zainab Faraj - who survived a triple-tap Israeli strike -told us she and her colleague Amal Khalil were deliberately targeted for reporting on the ground.
They paid the ultimate price for documenting Israel’s actions inside the so-called “yellow zone,” now off-limits to the Lebanese state and its citizens.
Concretely, this means that with journalists and residents driven out, Israel is carrying out the systematic destruction of Lebanon’s border villages behind closed doors.
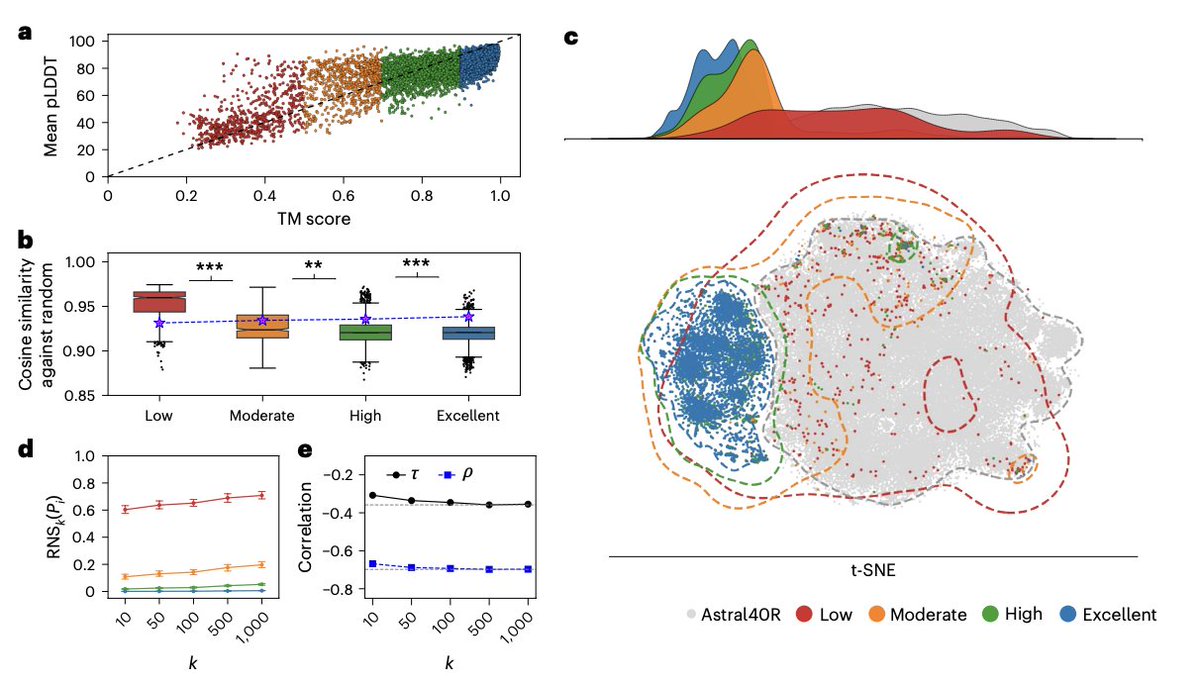
When a protein embedding is indistinguishable from noise
Protein language models have become the backbone of computational biology. Feed them an amino acid sequence, and they return a dense vector—a compact numerical fingerprint that downstream models use to predict function, structure, localization, or the effect of a mutation. The assumption, largely unquestioned, is that this fingerprint actually encodes meaningful biology.
Prabakaran and Bromberg challenge that assumption directly. They ask a deceptively simple question: how do you know whether a given embedding actually represents a protein—or whether it's just noise dressed up as a vector?
Their answer is the Random Neighbor Score (RNS). The idea is elegant: generate biologically meaningless sequences by randomly shuffling the residues of real proteins—preserving amino acid composition but destroying all evolutionarily meaningful interactions. Then, for each real protein, measure how many of its nearest neighbors in latent space are these random imposters. A high RNS means the model never learned to place that protein somewhere biologically meaningful.
Applied to ESM-2 and ProtT5 across thousands of proteins, RNS correlates strongly with structural prediction quality: proteins with poorly predicted structures have embeddings nearly indistinguishable from random sequences. Downstream tasks follow the same pattern—contact prediction precision drops roughly 40% for high-RNS proteins, and variant effect prediction falls to near chance. Most sobering: between 19% and 46% of the human proteome is underlearned by current models, depending on architecture. Intrinsically disordered regions fare especially poorly across all architectures tested.
RNS is model-agnostic and computationally cheap—around two minutes on GPU for 10,000 proteins—making it a practical prescreening step before any embedding-based inference.
For R&D teams that routinely use protein embeddings to prioritize variants, annotate novel sequences, or screen large libraries, this has immediate consequences. Running RNS before downstream inference flags proteins where predictions are unreliable, reducing the risk of propagating errors into expensive wet-lab campaigns. It also offers a principled way to identify gaps in model coverage—directly actionable for teams building or fine-tuning their own foundation models.
Paper: R. Prabakaran & Yana Bromberg, Nature Methods (2026) — CC BY-NC-ND 4.0 | https://t.co/JOblk7kT6R
Our group is hiring a postdoc to work on Digital Twin development for rare diseases. Collaborate with me and a fantastic interdisciplinary team! Deadline coming up this Sunday!
Our paper on the Protein Design Archive has just been published in @NatureBiotech! It describes our web application for exploring designed proteins (link ⬇️) and contains analysis of how they have changed over time. @MartaChronowska@mjstam@WoolfsonLab
https://t.co/7aa50oYRqQ
De Novo Design of Proteins for Autocatalytic Isopeptide Bond Formation | Journal of the American Chemical Society @UCSDChemBiochem https://t.co/0vWAoqiVv1
🙏 Special thanks to my amazing collaborators: @IoannisRiziotis , Rob Barringer, @andreeva789bg and to my supervisor @Alexbateman1 for their invaluable contributions to this work!
📢 Just published! Introducing Isopeptor: a computational tool for detecting intramolecular isopeptide bonds in protein structures with high precision and recall.
https://t.co/CYbH7tgoKg
💻 Available as a Python package for easy integration into bioinformatics workflows, and accessible via Google Colab for everyone: https://t.co/ybeOU0b6rz
Check out this article about my research at @emblebi where I focus on protein modelling and on the study of fibrillar adhesins. Recently, we developed a method to detect isopeptide bonds, a key stabilizing feature in bacterial proteins. Big thanks to @oanastroe123!!👇
Meet Francesco Costa @atsoCF 🇮🇹 – a PhD student at EMBL-EBI, focusing on protein science.
Francesco is fascinated by protein design, and his recent work helped ‘rescue’ low-confidence #AlphaFold protein structure predictions.
https://t.co/KkAkxqKAQM
#PhD
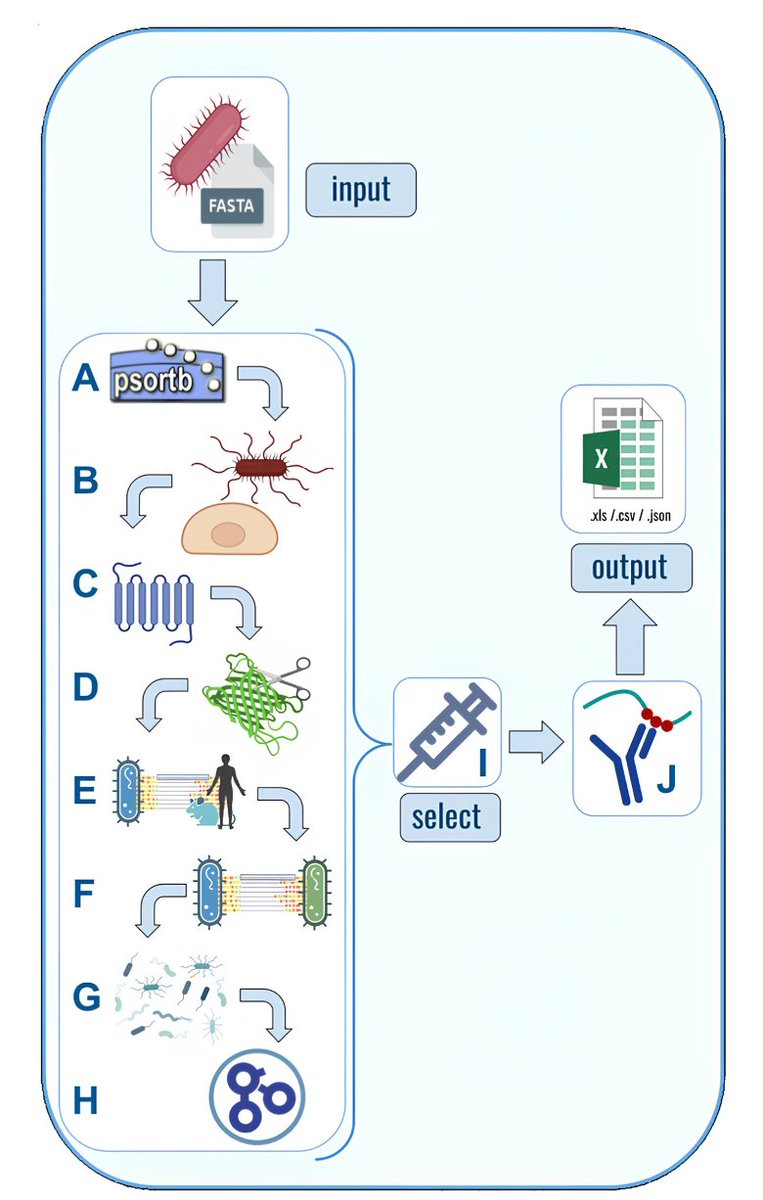
NERVE 2.0: Boosting the New Enhanced Reverse Vaccinology Environment via Artificial Intelligence and a User-Friendly Web Interface
1. NERVE 2.0 introduces updates to reverse vaccinology with AI-powered tools and a web interface, aiming to simplify vaccine candidate identification and analysis for diverse users.
2. New modules like ESPAAN predict adhesins with improved accuracy, while Virulent expands the search to virulence factors, addressing broader vaccine development needs.
3. Loop-Razor recovers extracellular regions of transmembrane proteins, allowing researchers to explore potential candidates often overlooked in previous pipelines.
4. Epitope Prediction targets epitopes with broad population coverage, utilizing linear HLA alleles to streamline immunogenicity assessments.
5. The redesigned web platform improves usability with modular options, adjustable parameters, and data storage, making it accessible to researchers with varying levels of bioinformatics expertise.
6. Benchmarking results show NERVE 2.0 performing better than its predecessor and tools like Vaxign2 and VaxiJen, demonstrating reliable enrichment in identifying bacterial protective antigens.
7. With both web-based and standalone versions, NERVE 2.0 supports flexible workflows for comprehensive reverse vaccinology analyses.
@Francescop966
💻Code: https://t.co/f0oRG3e9dM
📜Paper: https://t.co/MXa2Av1SCw
#ReverseVaccinology #Bioinformatics #VaccineDevelopment #MachineLearning #AI
Prediction confidence scores help #AlphaFold users gauge the reliability of protein structure predictions.
But things get more challenging for protein families.
This new method helps improve low confidence predictions within protein families.
https://t.co/KPhXxUp6Ta
Excited to announce our latest publication: “Keeping it in the family: Using protein family templates to rescue low confidence AlphaFold2 models” where we explore plDDT variability in #AF2 models of @PfamDB domains. https://t.co/qi8ZAQi1B3 1/7
Our findings have important implications for improving structure predictions, especially for proteins from organisms with limited representation in sequence databases or for rapidly evolving taxa. 6/7