We are looking for a Doctoral Candidate with a #bioinformatics background to use metagenomics to study microbial pathogens in non-clinical niches as part of #SPOILCONTROL @MSCActions DN DC5 - Pathogenic Flora https://t.co/GBNHsqjhfW
A new spatial transcriptomics approach eliminates the need for time-intensive imaging with specialized equipment, making the technique accessible to more researchers around the world. https://t.co/Yvki9deWE8
Identifying 14-3-3 interactome binding sites with deep learning
1. The study introduces a deep learning framework designed to predict protein binding sites to the 14-3-3 family, central hub proteins involved in various cellular signaling processes.
2. The researchers created an ensemble model combining multiple deep learning architectures, achieving 75% balanced accuracy in predicting 14-3-3 binding sites, even for challenging intrinsically disordered proteins.
3. This model was applied to 300 sequences of medically relevant proteins, including those linked to diseases like Alzheimer’s (Tau) and cancer (Myc, p53), identifying promising binding sites for further validation.
4. Experimental validation confirmed the model's predictive power, with five out of eight top predictions showing significant binding to 14-3-3 proteins, demonstrating the model's real-world applicability.
5. The study further supports the integration of deep learning with experimental methods such as X-ray crystallography and molecular dynamics to gain insights into protein-protein interactions and their structural dynamics.
6. The model was tested on various 14-3-3 client proteins like FOXO3, BAD, and Tau, with the top predictions providing new avenues for drug design targeting these key interaction sites.
7. By making the model accessible online (https://t.co/8AQrEt13Wg), the authors offer a tool for researchers to explore 14-3-3 interactions in their own studies, advancing our understanding of cellular signaling mechanisms.
💻Code: https://t.co/8AQrEt13Wg
📜Paper: https://t.co/IDk6p2nwUP
#DeepLearning #Proteomics #Bioinformatics #ProteinInteractions #Phosphorylation #Alzheimers #Cancer
A 100-billion-parameter unified transformer for interpreting and generating protein sequences
Proteins underpin vital processes in biology, from cellular signaling to enzyme catalysis. Their amino acid sequences determine complex structural and functional properties, making it essential to capture the informational patterns embedded in these chains. In recent years, large-scale computational methods have emerged to learn meaningful representations of protein sequences, assisting researchers in areas such as protein structure prediction, activity engineering, and novel sequence generation.
Chen et al. introduce xTrimoPGLM, which unifies two core learning strategies—masking random tokens for bi-directional context encoding and autoregressively predicting continuous spans—within a single 100-billion-parameter transformer model trained on one trillion amino acids. The authors leverage a dual-stage schedule that first emphasizes masked language modeling, then integrates both the masked and autoregressive objectives. This enables the transformer to excel in residue-level understanding while generating biologically plausible sequences. Quantitatively, the model achieves perplexities as low as 6.70 on out-of-distribution tests, surpassing similarly large baselines. It also scales according to a power-law relationship, showing steady gains in representation quality when trained on increasing amounts of data.
The researchers demonstrate that xTrimoPGLM substantially outperforms prior large-scale protein language models across 18 tasks spanning structural classification, stability prediction, and molecular interactions. The model supports rapid folding calculations, with a specialized module yielding high TM-scores on challenging benchmarks. It also generates de novo sequences with diverse structures, which suggests strong potential in customizable protein engineering. By uniting advanced bi-directional encoding with autoregressive generation, this framework offers a versatile platform to advance both understanding and design of proteins at scale.
Paper: https://t.co/gJ3y8Lw6Ad
🚀 Update! Our latest Pinal bioRxiv now includes wet lab results. More proteins with diverse text prompt on the way.
Design proteins with just text. Everyone can do protein design!
Demo: https://t.co/4XxGI0Hp9x
paper: https://t.co/mXSygLEa0w
GitHub: https://t.co/mm8mynEuxL
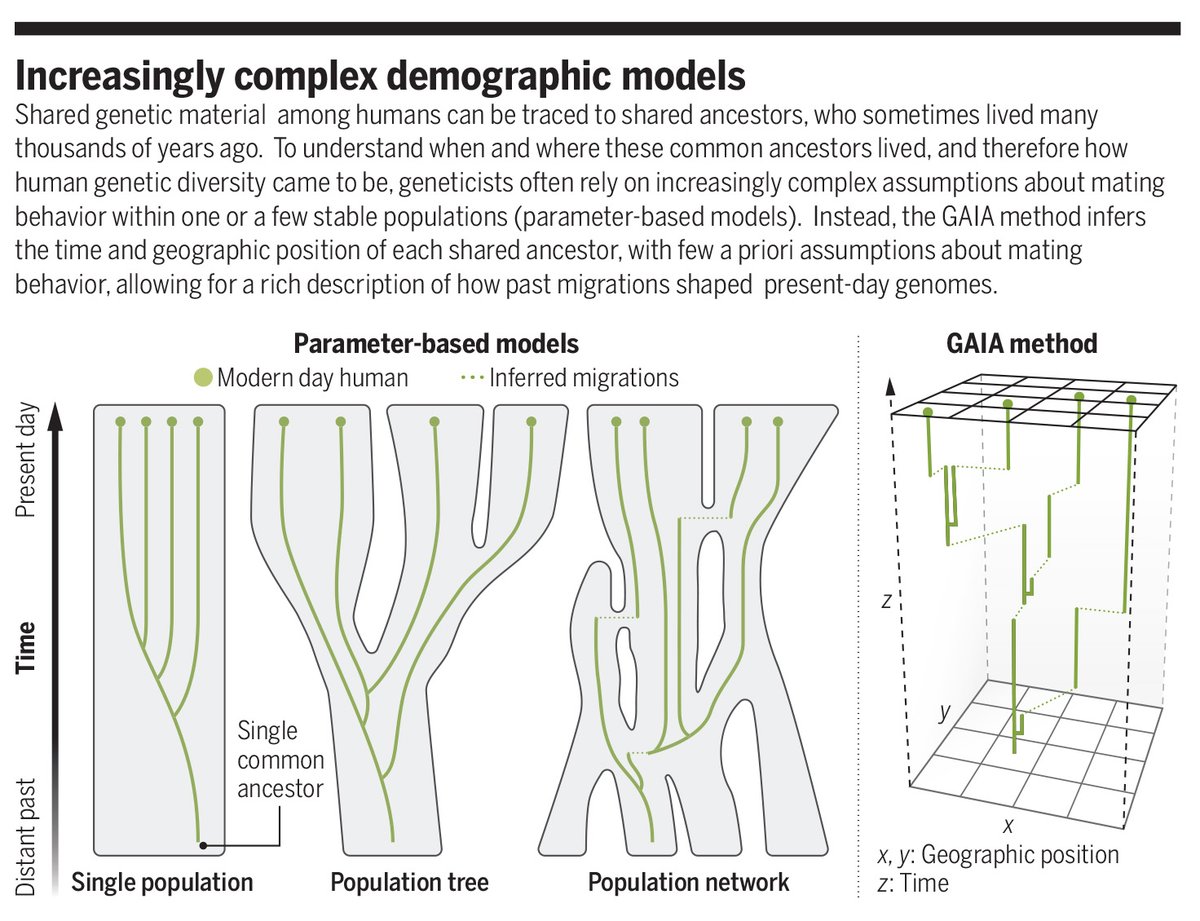
In a new Science study, researchers introduce GAIA, a statistical approach that seeks to learn the geographic position of every genetic ancestor of individuals included in a genome dataset.
Learn more ⬇️
📄: https://t.co/cD9oSZuEoE
#SciencePerspective: https://t.co/ikQfv84nYq
Entropy is one of those formulas that many of us learn, swallow whole, and even use regularly without really understanding.
(E.g., where does that “log” come from? Are there other possible formulas?)
Yet there's an intuitive & almost inevitable way to arrive at this expression.
We are incredibly small and insignificant. But we are intrepidly exploring the cosmos with technology made with human hands and human brains. First #JWST deep field shows thousands of galaxies and gravitational lensing. Things with lens flares = stars that are much closer to us.
The first image from the Webb Space Telescope represents a historic moment for science and technology. For astronomy and space exploration.
And for America and all humanity.