🚀 Excited to share our new work: Absolute Stability Predictor!
📊: https://t.co/gtgQjPRAX6
Built the MGnify Stability Dataset (1.8M+ measurements) and developed stability prediction models, together with @grocklin, @KotaroTsuboyama, @sokrypton, and teams.
1/ Excited to share our new paper in Science: “Toward life with a 19-amino acid alphabet through generative artificial intelligence design.” @ColumbiaSysBio@ColumbiaBME@Columbia
https://t.co/ZT3Ygw9tiG 🦠🧬🛠️🖥️💥
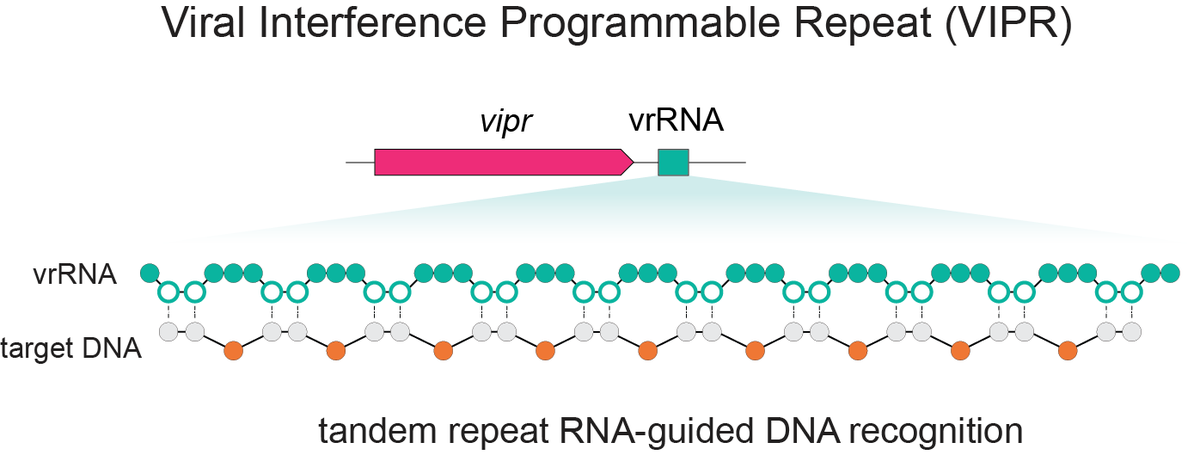
Excited to share our discovery of a new programmable RNA-guided DNA-targeting system hiding inside bacteriophages that predates CRISPR.
We call it VIPR (Viral Interference Programmable Repeat), and it uses an entirely new logic to find its targets.
Thread + link below.
🥳 Huge preprint 🔔 Today we share something our group has been working toward for a long time, led by
@LucasMoriniere We asked can we predict which receptor a phage targets from its genome sequence alone? For most phages, we couldn’t. So Lucas set out to do something crazy!
Big news! Starting my lab as Ramón y Cajal PI at @IBVF_Sevilla in Seville 🌞, Spain, bridging microbial ecology, photosynthesis & plant biotech: from metagenomic and AI analyses 💻 to experiments in microbes & plants 🌊🧬🌱 Looking for PhD students & postdocs. DM or share!
Happy to see this project that started during my PhD come to completion! Beautiful work by @AlexanderRotsch and team solving the structure of the influenza polymerase/elongating RNA polymerase II complex. Congrats to everyone for the amazing story! https://t.co/CwIKpksfaR
Predicting protein-protein interactions (PPIs) at proteome scale can take months with co-folding models due to massive all-vs-all comparisons required.
We are excited to announce FlashPPI, a contrastive model that predicts proteome wide physical interfaces in minutes. 1/🧵
We identify conjugative megaplasmids in the human gut that embed intact temperate prophage genomes and may enable cross-lineage viral dissemination.
We propose a plasmid-mediated route for temperate phage spread.
https://t.co/WlIyRNCXHO
Introducing DroPE: Extending the Context of Pretrained LLMs by Dropping Their Positional Embeddings
https://t.co/brpejkosWR
We are releasing a new method called DroPE to extend the context length of pretrained LLMs without the massive compute costs usually associated with long-context fine-tuning.
The core insight of this work challenges a fundamental assumption in Transformer architecture. We discovered that explicit positional embeddings like RoPE are critical for training convergence but eventually become the primary bottleneck preventing models from generalizing to longer sequences.
Our solution is radically simple: We treat positional embeddings as a temporary training scaffold rather than a permanent architectural necessity.
Real-world workflows like reviewing massive code diffs or analyzing legal contracts require context windows that break standard pretrained models. While models without positional embeddings (NoPE) generalize better to these unseen lengths, they are notoriously unstable to train from scratch.
Here, we achieve the best of both worlds by using embeddings to ensure stability during pretraining and then dropping them to unlock length extrapolation during inference. Our approach unlocks seamless zero-shot context extension without any expensive long-context training.
We demonstrated this on a range of off-the-shelf open-source LLMs. In our tests, recalibrating any model with DroPE requires less than 1% of the original pretraining budget, yet it significantly outperforms established methods on challenging benchmarks like LongBench and RULER.
We have released the code and the full paper to encourage the community to rethink the role of positional encodings in modern LLMs.
Paper: https://t.co/Fp5IJS4LIC
Code: https://t.co/Wvea7tQ5Ay
I'm super excited to announce the first preprint of my PhD, together with Chenxi Ou and
@sokrypton!
ML has revolutionized protein modeling, but key challenges remain. For example, we can't predict complicated protein structures without MSAs, which limits what we can design.
An energy-based model of protein conformational space can be used to predict structure from sequence, sample from the conformational landscape, rank structures, and predict mutation effects.
@jamesproney@sokrypton
Excited to share my recent work, CIRPIN 🐍 with @sokrypton and @MazAbulnaga on the discovery of thousands of naturally occurring circularly permuted protein domains
Excited to share this work with @yoakiyama@ChoYehlin@jajoosam@sokrypton
We find that protein language models trained solely on individual protein sequences, implicitly learn the interface contacts of homo-oligomeric assemblies! As the model scales up, more interface signals pop up.
However, we also notice some proteins PDB database marked as homo-oligomers are missing these interface signals. This took us on the journey to investigate the emergence and absence of homo-oligomeric contacts in pLMs.
https://t.co/sdxQTFgDBx
We're thrilled to announce SeqHub, an AI-enabled platform for biological sequence analysis. SeqHub brings together sequence search, genome annotation, and data sharing in one place.
I dreamed of a single place where I could learn everything about my sequences. Today, a much more refined version of this dream takes form with https://t.co/1l5O5cP3tE, built by an incredible team at @tatta_bio. Our goal is to make sequence interpretation more intuitive and collaborative for everyone working with biological sequences.
Currently, SeqHub is optimized for microbial protein and genome analysis. As we expand beyond microbial data, we'd love your feedback to help shape what comes next. I'm deeply grateful to our team at Tatta Bio, and to our collaborators and funders, for making this vision a reality.
Check it out at https://t.co/vXbVqe507X!
Today in @Nature , we highlight how a cousin of CRISPR-Cas10, mCpol, establishes an evolutionary trap in anti-phage immune systems.
Check out Erin Doherty's and my work from Doudna lab here:
https://t.co/ORz0t5CMvX
Welcome to the age of generative genome design!
In 1977, Sanger et al. sequenced the first genome—of phage ΦX174.
Today, led by @samuelhking, we report the first AI-generated genomes. Using ΦX174 as a template, we made novel, high-fitness phages with genome language models. 🧵
Excited to share work with @ZhidianZ, Milot Mirdita, Martin Steinegger, and @sokrypton
https://t.co/pkWeguhQ4l
TLDR: We introduce MSA Pairformer, a 111M parameter protein language model that challenges the scaling paradigm in self-supervised protein language modeling
🧵