LinkLlama: Enabling Large Language Model for Chemically Reasonable Linker Design
1. LinkLlama reframes fragment linking as instruction-following SMILES generation with explicit geometric constraints (fragment anchor distance in Å and angle in degrees) plus optional property bands—aiming to avoid the strained, non-drug-like linkers often produced by purely 3D generative methods.
2. The key design choice is “alignment-by-design”: instead of reinforcement learning loops, the model is supervised fine-tuned to internalize medicinal-chemistry constraints directly, then steered at inference time via natural-language prompts.
3. Training data comes from a large, curated ChEMBL36 pipeline: ~2.67M cleaned drug-like molecules are fragmented (two cuts via RDKit MMPA) into 8.3M fragment–linker–fragment triplets (256k unique linkers), with strict rules to keep linkers realistic (e.g., cuts on acyclic single bonds incident to neutral sp3 carbons; minimum sizes; linker not dominating fragments).
4. “Chemical reasonability” is operationalized with a stringent 5-part filter set: linker-level checks (bridgehead/overly complex ring structures; uncommon ring systems <100 occurrences in ChEMBL) and molecule-level checks (iMiner undesirable SMARTS, PAINS, Brenk). A molecule is “reasonable” only if it passes all five.
5. The model is Meta Llama-3.2-1B-Instruct fine-tuned with LoRA on 1.5M rebalanced examples (Cap50 per-linker frequency cap to reduce memorization of common motifs like amides). Training is lightweight (reported ~6.5 hours on 4×A100), emphasizing practicality.
6. A notable modeling twist: the output is a JSON containing both the proposed linker SMILES and a “reasoning” trace that explicitly reports pass/fail status for the same chemical filters—teaching the model to co-generate structures and their medicinal-chemistry compliance.
7. On standard ZINC benchmarks (random 1k + hard 1k), LinkLlama achieves near-perfect validity (~99.9%) and competitive uniqueness, while substantially increasing chemically reasonable designs: reasonability rises to ~73% (random) and ~87% (hard), vs ~44% for DeLinker and ~25–31% for DiffLinker.
8. 3D evaluation shows a key tradeoff improvement: fragment pose agreement (RMSD) is broadly comparable across methods, but LinkLlama’s MMFF strain proxy (ΔE) avoids the heavy tail seen in the 3D diffusion baseline, suggesting fewer distorted/high-strain linker geometries even though LinkLlama generates in SMILES space.
9. Prompt-conditioned control is a central capability: on ZINC hard 1k, LinkLlama can satisfy combined constraints (e.g., ring-containing + Lipinski Ro5 + minimum linker size/rotatable bonds + “reasonable”) with success rates in the ~40% range, while baselines and unconditional sampling collapse to near-zero under the strict joint constraints.
10. Prospective-style case studies extend beyond benchmarks: scaffold hopping on mineralocorticoid receptor (PDB 6L88) yields alternative heterocyclic cores with stable 200 ns MD behavior; PROTAC linker redesign on BRD4–VHL (PDB 6SIS) generates linear linker alternatives to a macrocyclic architecture, with MD suggesting comparable or improved ternary complex stability.
💻Code: https://t.co/mgQMHDjfzR
📜Paper: https://t.co/pynfL0eEe0
#ComputationalChemistry #DrugDiscovery #FBDD #GenerativeAI #LLM #Cheminformatics #PROTAC #MolecularDesign #MachineLearning
Kinconfbench: A curated benchmark for cofolding models on kinase conformational states
1 KinConfBench is introduced to evaluate whether protein–ligand cofolding models recover ligand-induced kinase conformational states (active/inactive microstates), not just plausible folds or good-looking ligand poses.
2 The benchmark contains 2,225 curated, high-quality human kinase chains (1,420 unique systems: 43 apo targets and 1,377 holo complexes), derived from a UniProt-centric PDB query (snapshot 2025-08-01) and filtered for motif completeness and structure quality.
3 Each kinase chain is annotated with KinCoRe, using eight categorical labels capturing mechanistically relevant features (DFG spatial/dihedral states, αC-helix orientation, salt bridge, activation loop N/C labels, regulatory spine, and an overall activity label). A prediction is “state-correct” only if all eight labels match experiment.
4 Three open-source cofolding models are benchmarked under standard inference pipelines: Boltz-2, Chai-1, and Protenix. For each target, N=20 samples are generated and ranked by confidence; outputs are re-annotated with KinCoRe to score conformational fidelity.
5 A key finding: common ligand-centric geometric success criteria (global lDDT-Cα, pocket lDDT-PLI, ligand RMSD) correlate only weakly with correct kinase conformational state. Models can place ligands well while missing the induced-fit regulatory layout.
6 On a filtered subset where all models can generate at least one geometrically valid pose (950 systems), conformational classification accuracy is ~65–75% across models, but ensemble behavior is strongly bimodal: many targets are either “all right” or “all wrong” across the 20 samples, suggesting mode collapse rather than calibrated uncertainty.
7 A representative failure mode is highlighted (MAP4K1, PDB 7M0M): all models achieve essentially correct ligand placement yet miss the experimentally observed activation-loop/DFG-related induced-fit configuration, demonstrating how ligand RMSD can mask protein-state errors.
8 Even when models hit the correct KinCoRe state, their within-state ensemble diversity is very low (typical ~0.1 Å dispersion in key distances and <5° in key torsions), far below fluctuations expected for thermalized protein ensembles, implying limited sampling of induced-fit motions.
9 A paired apo→holo generalization test shows prevalent “apo-drift”: when ligands should induce a different KinCoRe state than apo, models frequently revert to apo-like conformations, and this worsens for holo states appearing after each model’s training cutoff—consistent with memorization of common apo baselines rather than robust ligand-modulated conformational reasoning.
10 The work argues that next-generation SBDD evaluation should prioritize protein conformational correctness and ligand-induced conformational diversity, complementing standard pose metrics; KinConfBench is positioned as a step toward dynamical/state-aware benchmarking for cofolding models.
💻Code: https://t.co/x6wklnqOLI
📜Paper: https://t.co/jicbdwD9yT
#computationalbiology #bioinformatics #structuralbiology #kinase #drugdiscovery #SBDD #proteinfolding #AI4Science #machinelearning #benchmarking
❓How can we build AI agents that do what scientists actually do? Is scientific discovery merely a search problem?
🚀 Meet SAGA: Scientific Autonomous Goal-evolving Agents. Five discovery tasks across chemistry, biology & materials science, with wet-lab validation.
The undergraduate work of @kunyangsun and spearheaded by Matthew Du is finally out. It shows that delocalization of dark states in multimode cavities is surprisingly qualitatively different to the case of single mode cavities.
https://t.co/FrsflLlUjU
This is my first attempt to do interesting things with LLM! The code is still being finalized and will be out soon. I'm currently at #ACSSpring2025 and if you are interested in chatting, connect with me here (https://t.co/QbmxfvIjGA)! More discussions about this work is coming.
SynLlama: Generating Synthesizable Molecules and Their Analogs with Large Language Models
- Traditional generative models for drug discovery often fail due to poor synthesizability of generated molecules. SynLlama addresses this by fine-tuning Llama-3 models to generate full synthetic pathways using accessible building blocks and robust reaction templates.
- Unlike other methods that require extensive reaction data, SynLlama achieves high performance with significantly less data while maintaining synthetic feasibility, making it a powerful tool for drug discovery.
- A key feature of SynLlama is its ability to propose not only target molecules but also their synthetic analogs, allowing medicinal chemists to explore viable alternatives with similar pharmacological properties.
- The model learns to generate synthetic pathways by mapping molecules to sequences of building blocks and chemical reactions, outperforming existing synthesis planning models in target molecule reconstruction and analog similarity.
- SynLlama expands the chemical space beyond predefined training data, effectively generalizing to new building blocks and synthetic routes, increasing its utility for real-world drug discovery applications.
- Experimental validation and benchmarking against SynNet, ChemProjector, and Synformer show that SynLlama achieves competitive or superior performance while requiring far fewer training examples.
- The model successfully aids in tasks like hit expansion and de novo molecule generation by improving synthesizability without compromising biological activity, as demonstrated in case studies with SARS-CoV-2 main protease inhibitors.
- With its ability to interface with other generative AI models, SynLlama serves as a bridge between theoretical drug design and practical chemical synthesis, making AI-driven drug discovery more actionable.
💻Code: https://t.co/2eaikymx5I
📜Paper: https://t.co/Y9WHjetGiW
#AI #DrugDiscovery #MachineLearning #ComputationalBiology #SyntheticChemistry #LLM
Existing AI benchmarks are becoming increasingly saturated.
Partnering with @scale_AI, we created Humanity's Last Exam, a new benchmark that tests the capabilities of models at the frontier of human knowledge and reasoning.
The results are out today:
https://t.co/JPC6dkeBKR
It is very sad to learn that Martin Karplus passed away on Dec 28 at age 94. He was a scholar and pioneering chemical physicist with great contributions in many areas including molecular dynamics. We interacted many times starting from my graduate time at Harvard. His legacy is reinforced by the large number of coworkers who have also done much to advance computational science.
PDBBind Optimization to Create a High-Quality Protein-Ligand Binding Dataset for Binding Affinity Prediction
• This paper presents PDBBind-Opt, an automated workflow designed to improve the PDBBind dataset by curating non-covalent protein-ligand binding complexes with enhanced data quality for binding affinity prediction.
• PDBBind-Opt addresses common structural issues in PDBBind, such as covalent binders, rare element ligands, and steric clashes, by filtering out problematic entries to ensure the accuracy and generalizability of scoring functions (SFs).
• The workflow includes modules for ligand and protein fixing, improving structural fidelity by correcting bond orders, protonation states, and missing atoms based on structural data from the RCSB PDB.
• PDBBind-Opt also refines the dataset by adding hydrogens in protein-ligand complexes, enhancing the accuracy of hydrogen bonding interactions for SFs.
• Additionally, PDBBind-Opt introduces BioLiP2-Opt, an independent dataset based on BioLiP2, providing a valuable resource for external benchmarking in structure-based drug discovery.
• The enhanced datasets offer a rigorous foundation for future scoring function development and are open-sourced to promote transparency and reproducibility in computational drug discovery.
@thg_lab@kunyangsun
💻Code: https://t.co/98NVQ4k5nk
📜Paper: https://t.co/FZjaXDVCBJ
#ProteinLigand #DrugDiscovery #BindingAffinity #Bioinformatics #PDBBind
Students of chemical physics, soft matter, biophysics, or anyone interested in understanding the visible world through a lens of its molecular constituents should check out Statistical Mechanics and Stochastic Thermodynamics, available for preorder https://t.co/Y761hftU3K
BREAKING NEWS
The Royal Swedish Academy of Sciences has decided to award the 2024 #NobelPrize in Chemistry with one half to David Baker “for computational protein design” and the other half jointly to Demis Hassabis and John M. Jumper “for protein structure prediction.”
SmileyLlama, a fine-tuned Chemical Language Model to design molecules from properties specified in the prompt. An SFT+DPO model on par with other pure CLM's, but built with Axolotl.
Also, big thanks to Prof. Bannister @UFScripps to offer suggestions from a med chem perspective! We are looking for presenting this work at the Neurlps workshop and are open for any potential recommendations! (4/4)
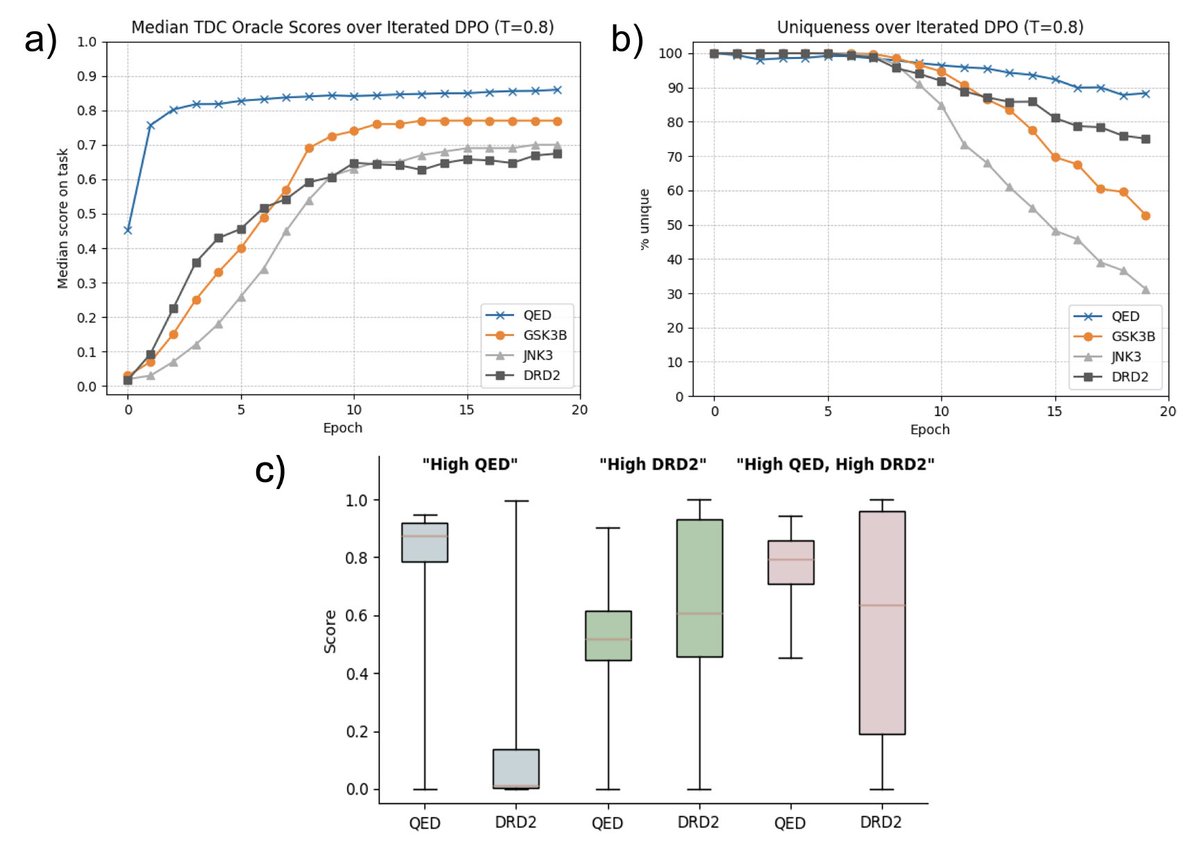
Fresh out of the arxiv! In this work, we (Joe, @dyushag , Dorian, and myself @thg_lab ) used supervised fine tuning (SFT) and direct preference optimization (DPO) to modify Llama 3.1 into a chemical language model. See more details here: https://t.co/NdWGtpsABl (1/4)
Another exciting finding is that SmileyLlama could implicitly leverage the idea of multi-objective optimization. We did four individual DPO runs on the same SmileyLlama model for each @ProjectTDC oracle functions, and it is able to optimize on 2 or more properties! (3/4)
🚀Join us at #IROS2024 for the "Equivariant Robotics: The Role of Symmetry Across Perception, Estimation, and Control" workshop!
✨We welcome the contribution of short papers / extended abstracts.
🌐Check out our website: https://t.co/Fr6NONRBGx
#Robotics#AI#symmetry