David Baker's lab (Institute for Protein Design) just designed genetically encoded miniproteins that directly control kinase activity — a target class notoriously hard to hit precisely because kinase catalytic domains are so structurally conserved that most inhibitors leak onto off-target kinases.
96 designs built to bind and stabilize distinct conformational states of FAK (focal adhesion kinase), targeting the kinase domain itself rather than an allosteric side pocket. 33 modulated activity. The 4 most potent: two inhibit FAK at low-nanomolar IC50, two potentiate it by more than 2×.
Expressed in living cells, the designs preserved the same inhibitory/activating effects seen in vitro — these rewire signaling inside real cells, not just in a test tube. Then the team exploited kinase structural similarity to redesign the FAK inhibitors into Src kinase inhibitors — a blueprint for genetically encoded, selective control across the entire kinase family.
https://t.co/oZJ6GcLjO2
Have you ever wanted to get into de novo protein design, but didn't know where to start?
Or are you just tired of writing messy one-off scripts to glue together RFDiffusion, ProteinMPNN, and structure prediction tools (ColabFold, AF3, Boltz2)?
Then we have a treat for you!
I've been really impressed with ESMFold2's design capabilities.
To make it easier to run locally, as well as to support structural templates, multichain targets, hotspots, Protenix-v2 validation, and automated MSA handling, I created this project:
https://t.co/EUTIkpJaUA
Affinity Fine-Tuning of Boltz-2: An Open Framework for Protein-Ligand Potency Prediction in Drug Discovery
Boltz-2のリガンド親和性予測のファインチューニングフレームワーク
固有の実験データでアフィニティ予測部分のみ最適化
https://t.co/T5htMQi0bP
https://t.co/41SAJMWtJq
I am happy to share a review I recently wrote on the design of peptide binders. It gives an overview of experimentally validated tools and discusses the challenges of why peptide design is more difficult than the design of classical protein binders.
https://t.co/vqShmqiJES
The next frontier in protein design will not be defined by structure alone, but by the capacity to engineer motion as a first-class principle of function. This is because dynamics is where the real biology lives.
Foundational work by Karplus, Levitt & Warshel made clear that chemistry cannot be understood without motion, mechanism, and scale. Gō, Brooks & others showed that proteins possess characteristic collective motions - low-frequency normal modes that capture how whole molecules bend, breathe, and fluctuate. Frauenfelder then sharpened the picture further: proteins are not static objects occupying a single minimum, but dynamic ensembles traversing rugged energy landscapes.
And yet the modern AI revolution in protein science has been, above all, a revolution in structure. In our new paper in Matter, @_Bo_Ni and I ask a different question: not what structure will this sequence adopt? but what sequence will realize a prescribed pattern of motion?
VibeGen inverts the conventional design paradigm. Rather than treating dynamics as a consequence to be analyzed after the fact, it makes dynamics the design objective from the outset. Using a language diffusion model with two cooperating agents - a designer that proposes sequences and a predictor that critiques them against the target motion profile - the system converges on de novo proteins with tailored vibrational behavior.
One of the most intriguing results is a form of functional degeneracy - distinct sequences and distinct folds can satisfy the same target dynamical specification. For a given functional pattern of motion, evolution may have sampled only a small region of the physically realizable design space. The space of viable molecular mechanics may be far larger than the repertoire biology happened to discover.
We have made "vibe" into a cultural metaphor - something intuitive, affective, subjective. But at the molecular scale, vibe is not metaphor: It is physics. For a protein, the vibe is the pattern of motion itself; the fluctuations, resonances, and collective displacements that determine what the molecule can do.
Paper: https://t.co/StOpyqmfzY
Code: https://t.co/WbdRrQYWeW
Authors: Michael F. Chungyoun, Sreevarsha Puvada, Gabriel Au, Courtney Thomas, Britnie J. Carpentier, Jeffrey J. Gray
Acknowledgments: Sergey Lyskov, Sergey Ovchinnikov, Johns Hopkins students of 2023 540.614/414 Protein Structure Prediction course, and the Johns Hopkins Center for Teaching Excellence and Innovation - Instructional Enhancement Grant.
I received many requests to share materials from our undergraduate course “Machine Learning in Chemistry”
— here you go!
A preprint summarizing insights and lessons learned:
https://t.co/UcQbWet75n
A Jupyter Notebook Tutorial Gallery:
https://t.co/dcgBzsTTe6
🎁 Application fee waiver for all attendees!
📅 Oct 8 @ 10 AM ET
👉 Registration: https://t.co/7qusFJb8NO
For more details about our program, visit: https://t.co/GzzfPlOBwD
A few scientific cover arts I created four journals recently — made possible by @Blender 's ability to handle biological models like protein structures and CT scans.
#b3d
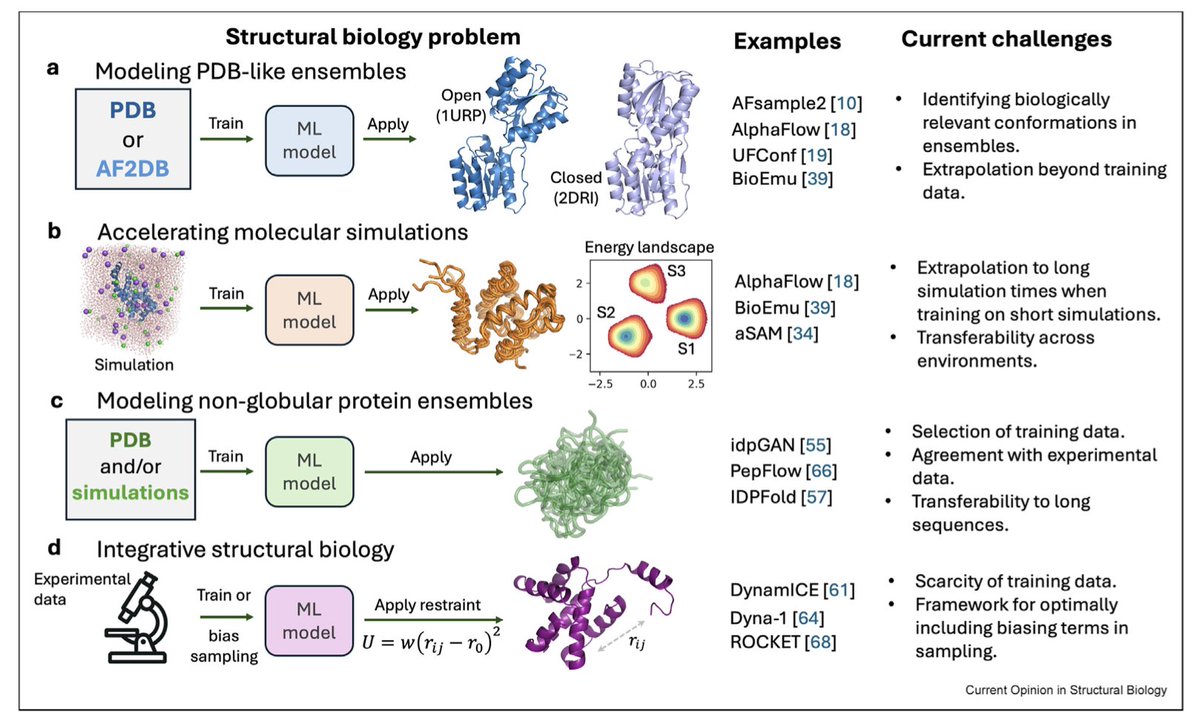
Generation of protein dynamics by machine learning
1. Machine learning, particularly generative models, is revolutionizing the prediction of protein dynamics by enabling the generation of structural ensembles beyond traditional simulations. This review highlights emerging approaches that capture protein dynamics in various forms, including PDB-like ensembles and acceleration of molecular simulations.
2. One significant innovation is the development of deep generative models (GMs) based on AlphaFold2, such as AlphaFlow and UFConf, which can generate multiple conformations from a single protein sequence. These models outperform traditional sampling methods in capturing PDB-like conformations.
3. The review emphasizes the importance of hybrid models that integrate experimental and simulation data. BioEmu, a diffusion model, demonstrates unprecedented performance in modeling both PDB and MD ensembles by leveraging a hybrid training strategy. This approach captures large and biologically significant conformational changes.
4. For non-globular proteins, especially intrinsically disordered regions (IDRs), ML methods are crucial for generating ensembles. Models like IDPFold and BioEmu show promise in capturing experimental observables of IDRs, such as chemical shifts and radius-of-gyration, using a combination of PDB structures and simulations.
5. The integration of experimental data directly into the generative process is another key advancement. Methods like DynamICE and DEERFold incorporate NMR and other experimental data during training, enhancing the accuracy of generated ensembles. This approach is essential for guiding ML models towards biologically relevant conformations.
6. Despite these advancements, challenges remain, including the transferability of models beyond training data and the generation of states with correct relative probabilities. The scarcity of long MD simulation datasets and the need for larger, more diverse training sets are also highlighted as critical areas for future work.
📜Paper: https://t.co/RlcASWEmx6
#MachineLearning #ProteinDynamics #StructuralBiology #GenerativeModels #Bioinformatics
A wonderful article on PepMLM by @xiaofei_lin in @GENbio! 🌟 Really captures how we think about sequence vs. structure and how, via experimental collaboration, we've gotten biologists (like Ray Truant, @DeLisaGroup, and @AguilarVirology) to believe in sequence-based design. 💚
Our first @DARPA program NODES is about to begin, come join us. Here is a glimpse of the broader vision.
"A to-do list for realizing the sequence-to-function paradigm of proteins" #currentopinionsinstructuralbiology
Despite AlphaFold's success, inferring function from sequence remains unsolved. This review lays out the roadmap to bridge this gap using:
🧠 Biophysical signatures
📉 Dimensionality reduction
🔬 Integrative modeling
💡 Machine learning beyond brute-force MD
Key insight? We don’t need all dynamics — just the right representations.
From MHC binding fingerprints to cell-scale simulations, this piece reframes the problem and offers a tractable path forward.
👉 Read more: https://t.co/ix5ai2H2CF
Check out NODES - https://t.co/iPPv311XsZ
🔍 #proteindynamics #AIinBiology #computationalbiology #structuralbio
NODES: Network of Optimal Dynamic Energy Signatures
program aims to develop a groundbreaking deep learning tool, informed by the principles of biophysics, that can analyze vast numbers of protein sequences and predict their biological functions by identifying telltale patterns of protein movement.
With this program, DARPA @DARPA seeks to characterize the function of potential biological threats within one hour, expanding threat assessment accuracy and speed exponentially. Speed is crucial for developing effective medical countermeasures for warfighters and for staying ahead of advancements in biotechnology that could lead to new and emerging biothreats.
Additionally, NODES will provide a secure system for classifying and monitoring biological threats, which is vital for intelligence agencies to enhance bio-surveillance.
https://t.co/rV9ZSVJXKs via @YouTube
the remarkable story behind this paper is best saved for another day when I can do it justice. for today, I thank the editor, who saw the potential in this work, and the reviewers, whose thorough evaluation of the paper helped improve it enormously
🚀 Super excited to share our new work, led by @SchnapkaV 🚀 bAIes, a Bayesian framework for generating accurate atomic-resolution ensembles of disordered & multi-domain proteins using #AlphaFold info! Now on @biorxivpreprint@ERC_Research https://t.co/VGKeTvs1SY
In a medical milestone, a customized base editor was developed, characterized in human and mouse cells, tested in mice, studied for safety in non-human primates, cleared by @US_FDA for clinical trial use, manufactured as a complex with an LNP, and dosed into a baby with a severe, rapidly progressing genetic disease... all in an astounding 7 months. Best of all, the infant patient shows apparent benefit. Congratulations to @kiranmusunuru, Rebecca Ahrens-Nicklas, and other team members for this heroic and inspiring effort, which has implications for the hundreds of millions of patients that suffer from thousands of genetic diseases.
https://t.co/wsgvvRYPVM
Students often get lost in complex frameworks of biology. Our animated lessons simplify concepts, saving time and clearing misconceptions!
Learn more at https://t.co/rZjuFXgP1n
#biology#3D#science#EdTech