presentation by @TBaharav today at @RECOMBconf on the statistics behind SPLASH, a method that can be used as a robust alternative to one of the most common statistical tests
backlogged on posting our new work in genomics! using RNA splicing as a special case of expansive, ultra-efficient and fully statistical analysis of RNA or DNA sequencing data. https://t.co/9Dwco1lmhW
Thrilled to share our new publication in PNAS on OASIS, an alternative to Pearson’s X² for analyzing contingency tables.
Made it to the front page! 1/ 7
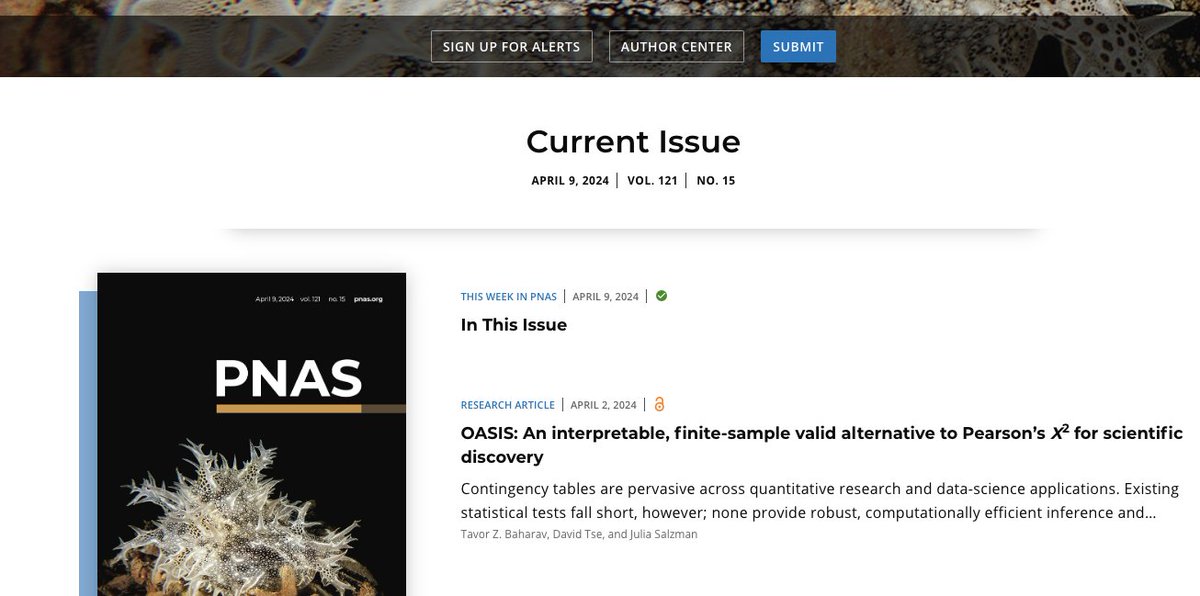
3. Development of NOMAD2, which enables reference-free biological discovery at unmatched scale and speed. In collaboration with @sdeorowicz and @marekkoki, led by @marekkoki and @roozbehdn, this package is publicly available on github: https://t.co/FFsvwH8siU
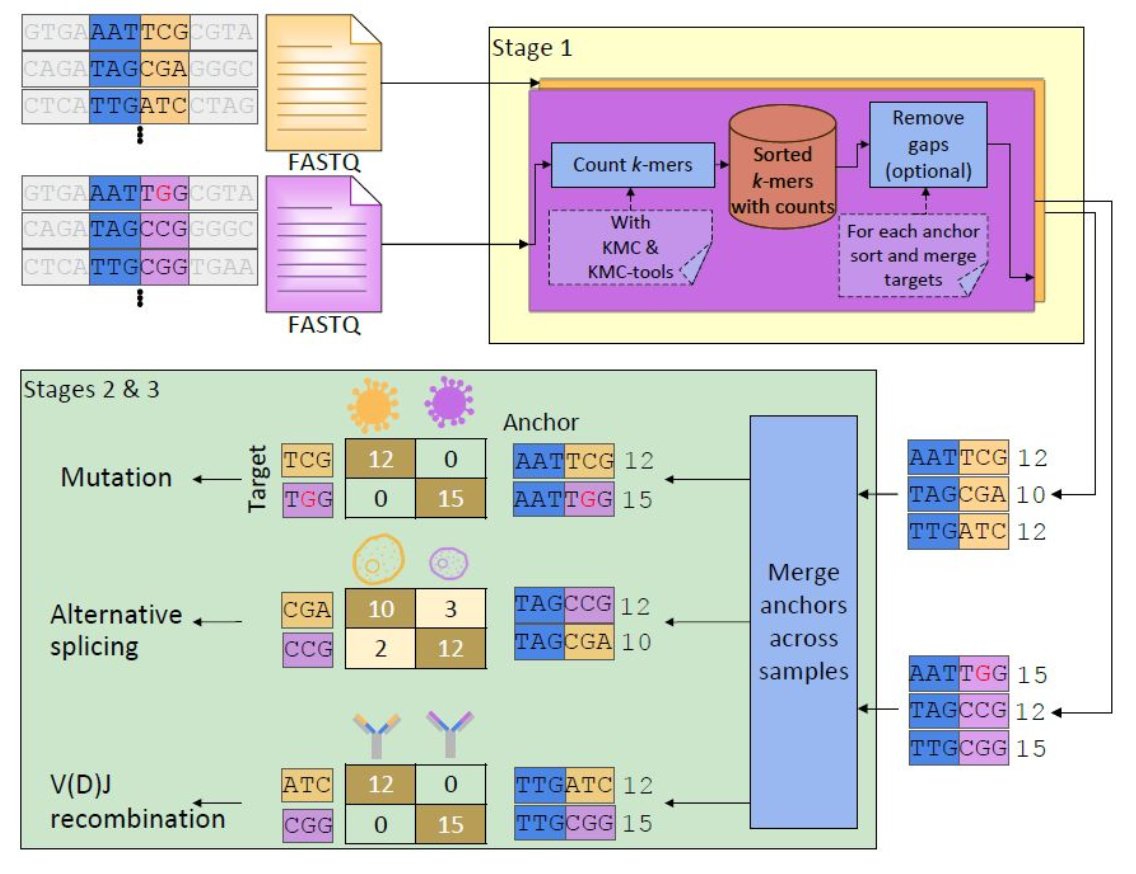
2. Design of a novel statistical test on contingency tables, OASIS, which provides interpretable and finite-sample valid rejection of the null, enabling reference-genome and metadata free strain detection in biological applications. Led by @TBaharav: https://t.co/GccCGdQirS
I can confirm that the NOMAD analysis method and the accompanying software is a game-changer for sequence analysis and discovery. Check this paper out! https://t.co/TewYcDIaHM
NOMAD2 is an unsupervised, reference-free, ultra-fast next generation of NOMAD, revealing new insights into cancer transcriptomes. Joint work with @salzmanlab. Many thanks to all for great collab!
https://t.co/x25YvXXhdh
NOMAD+ is a unified, highly efficient algorithm enabling unbiased discovery of an unprecedented breadth of RNA regulation and diversification in single cells through a new paradigm to analyze the transcriptome.
NOMAD+ discovers broad and new examples of transcript diversification in single cells, bypassing genome alignment and without requiring cell type metadata.
Very excited that ReadZS, a scRNA-seq pipeline for detection of RNA processing, is out in @GenomeBiology! This work was led by @ea_meyer, K Chaung, and @roozbehdn.
ReadZS, from @ea_meyer, Chaung, @roozbehdn & Salzman, for identifying RNA processing (splicing, alternative polyadenylation etc) in RNA-seq data, without the need for pre-existing annotations. It works on both 3'-enriched and non-enriched data https://t.co/HIHi2baC6F
Excited to share our new preprint NOMAD! We present a unifying statistical formulation for many fundamental problems in genome science and develop an efficient reference-free algorithm (NOMAD) to solve it. Work by Kaitlin Chaung, @TBaharav, and @RZheludev. https://t.co/2SFbqQ43Ia
Excited to announce that a project spearheaded between our lab and David Tse's has been selected for a @czbiohub Data Insights Award! Follow the link for more information.
https://t.co/BtlIKu0rRl
Excited to share DIVE is live on bioarxiv! DIVE is a novel reference-free approach for de novo discovery of diversity-generating and mobile genetic elements implemented as a publicly available python package (work led by @jordiatjhu).