gander is an R package that brings AI directly into RStudio or Posit.
Instead of switching between your IDE and a chat window, gander lets you ask questions or request code changes right inside your script. It automatically shares relevant context such as variable names, data types, and the surrounding code, so the model can provide precise answers without extra copy-pasting.
You can trigger it with a simple keyboard shortcut, choose from different AI models (OpenAI, Claude, or local ones), and control how much of your data is sent for context. In short, gander makes working with AI in RStudio smoother, faster, and smarter.
Take a look at the visualization below. It shows an example of how to use gander to create a ggplot2 graph. It’s taken from the package website: https://t.co/DbsZTbsmVK
In a recent Statistics Globe Hub module, you will learn step by step how to use gander for AI-assisted coding in R, write effective prompts, and apply it to real data science workflows.
The Statistics Globe Hub is an ongoing learning program focused on practical skills in statistics, data science, AI, and programming with R and Python.
More info about the Statistics Globe Hub: https://t.co/NA2b7UAXJ4
#AI #DataScience #RStats #Coding #Programming #DataAnalytics #statisticsglobehub
After 9 months of using NotebookLM, I can say it's the research tool that has revolutionized my workflow the most.
But only because I learned these 20 prompts.
Here's the complete system that turns 500 pages into clear answers in under an hour:
INSTEAD OF WATCHING NETFLIX TONIGHT.
Spend 1 hour with this.
Claude AI FULL COURSE that teaches you how to BUILD and AUTOMATE anything.
The people who watch this tonight will wake up tomorrow with a skill that most people will not have in 2 years.
The people who skip it will still be watching Netflix next year wondering why nothing in their life has changed.
Your call.
Most protein-protein interaction tools work on protein pairs. FlashPPI runs at proteome scale and now across two proteomes at once.
Upload any two datasets, get back a predicted interaction network spanning both.
BREAKTHOUGH: Google's AI can now read 1 million DNA letters at once.
Google's DeepMind has unveiled a revolutionary deep learning model, AlphaGenome, which can analyze long sequences of DNA with remarkable accuracy.
A new peer reviewed study published in Nature, AlphaGenome can process up to 1 million base pairs (1 megabase) in a single input, capturing long range genetic interactions that previous models could not.
The system predicts how single letter DNA changes affect gene expression, RNA splicing, and chromatin regulation across 11 genomic signals, even within the 98% of the human genome that does not code for proteins. In benchmark tests, AlphaGenome matched or outperformed previous state of the art models at identifying functionally important genetic variants.
By making large sections of the non-coding genome interpretable, AlphaGenome could significantly accelerate disease variant discovery, cancer research, and precision medicine, moving genomics from sequence reading toward functional understanding.
AlphaGenome: Decoding the dark matter of the genome with a unified deep learning model
More than 98% of human genetic variation lies outside protein-coding regions. These "non-coding" variants can disrupt gene regulation in remarkably diverse ways: altering chromatin accessibility, shifting 3D genome architecture, modifying splicing, or changing expression levels—often in tissue-specific patterns. Yet existing computational models face a fundamental trade-off: either they capture long-range regulatory interactions (like distant enhancers) but blur fine-scale features, or they achieve nucleotide resolution but miss distal context. And most specialize in a single modality, leaving users to stitch together predictions from many separate tools.
Žiga Avsec and coauthors at Google DeepMind present AlphaGenome, a model that sidesteps these trade-offs. It takes 1 megabase of DNA as input and predicts ~6,000 genome tracks—spanning gene expression, splicing (sites, usage, and junctions), chromatin accessibility, histone modifications, transcription factor binding, and 3D contact maps—at up to single-base-pair resolution.
The architecture combines a U-Net backbone with transformer blocks: convolutions capture local motifs essential for splice sites and TF footprints, while transformers model long-range dependencies like enhancer–promoter interactions. Training uses a two-stage approach—pretraining on experimental data followed by distillation from an ensemble of teachers using mutationally perturbed sequences—yielding a single model that scores variants across all modalities in one pass.
The results are striking: AlphaGenome achieves state-of-the-art performance on 25 of 26 variant effect prediction benchmarks, including a 25% improvement in predicting eQTL direction over the previous best model. It outperforms specialized models on their own tasks—beating SpliceAI-class methods on 6 of 7 splicing benchmarks and ChromBPNet on accessibility QTLs. Critically, the multimodal outputs enable mechanistic interpretation: for oncogenic mutations near the TAL1 gene in T-cell leukemia, AlphaGenome simultaneously predicts neo-enhancer formation (increased H3K27ac), chromatin opening, and elevated gene expression—recapitulating experimentally validated mechanisms.
This points toward a future where interpreting non-coding variation no longer requires assembling a patchwork of specialized models. A unified framework that jointly predicts molecular consequences across modalities could accelerate rare disease diagnostics, guide therapeutic oligonucleotide design, and help prioritize variants in GWAS loci—moving us closer to truly reading the regulatory code written in DNA.
Paper: https://t.co/3WzrnGNUSw
AlphaGenome is out in @nature today along with model weights! 🧬
📄 Paper: https://t.co/1fHzSPiY1x
💻 Weights: https://t.co/z6JWLT4Mpv
Getting here wasn’t a straight path. We sat down @googledeepmind to discuss the story behind the model, paper & API: https://t.co/cT8CiXfnxQ
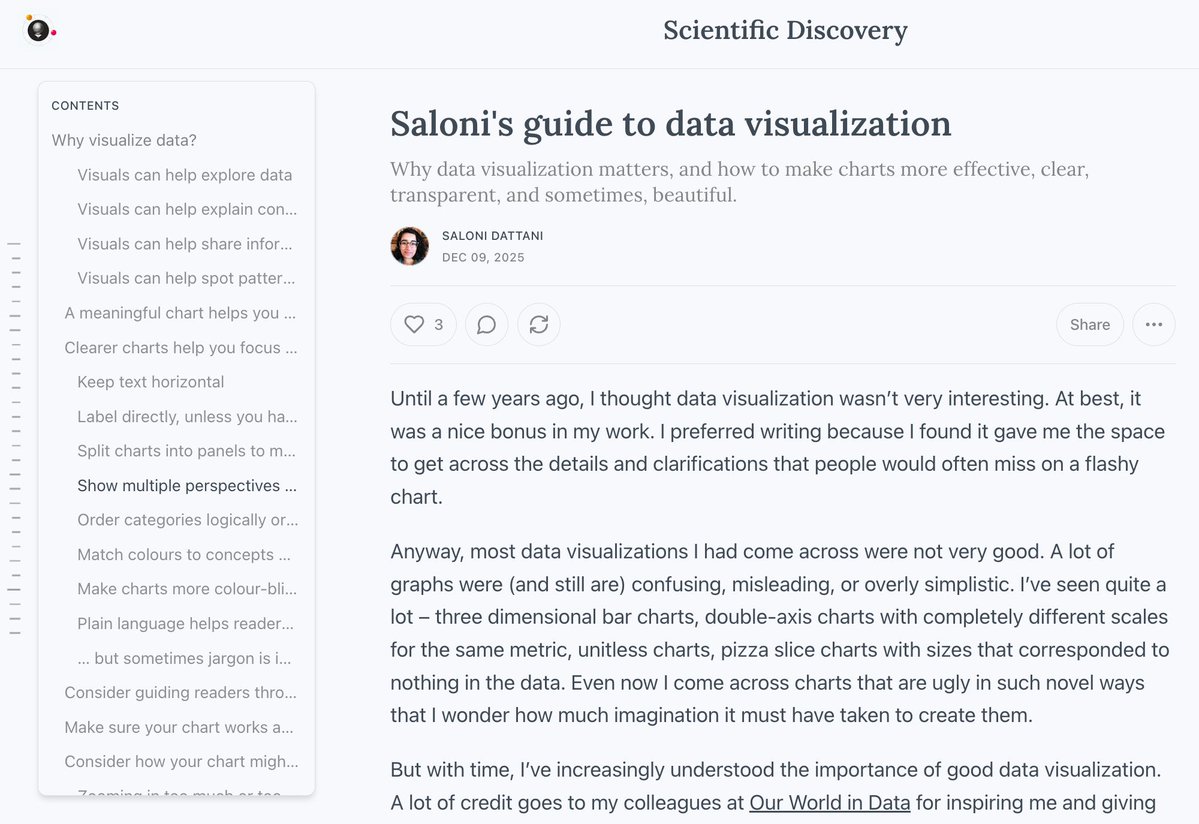
Big new blogpost!
My guide to data visualization, which includes a very long table of contents, tons of charts, and more.
--> Why data visualization matters and how to make charts more effective, clear, transparent, and sometimes, beautiful.
https://t.co/hDQhDL5rR1
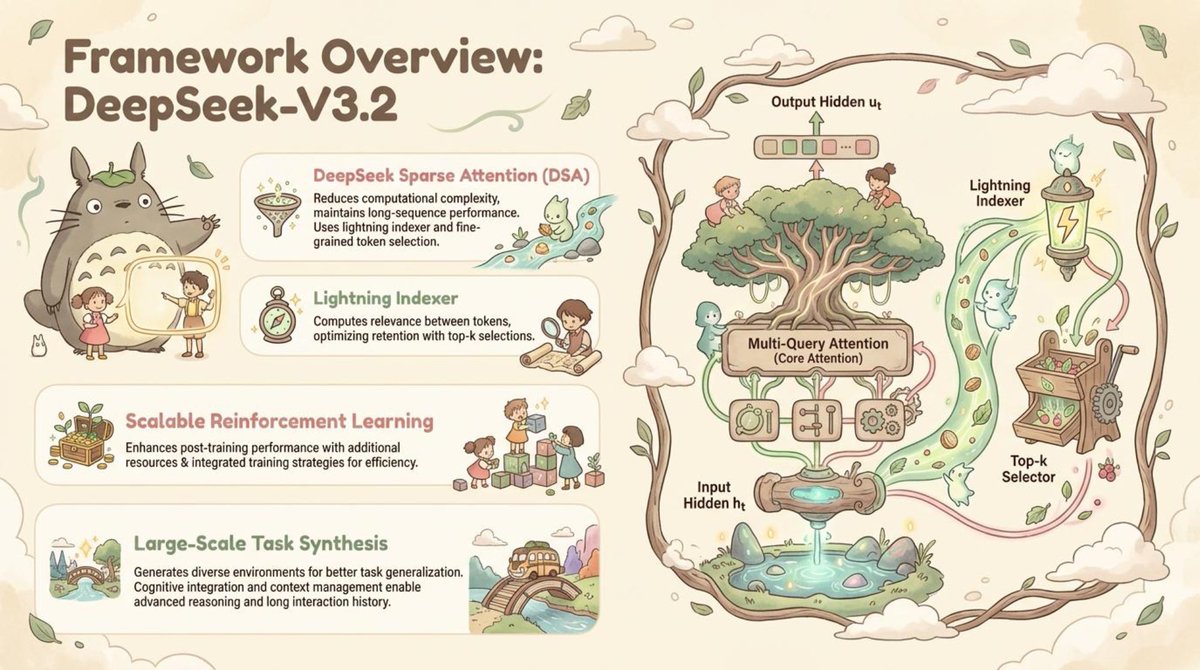
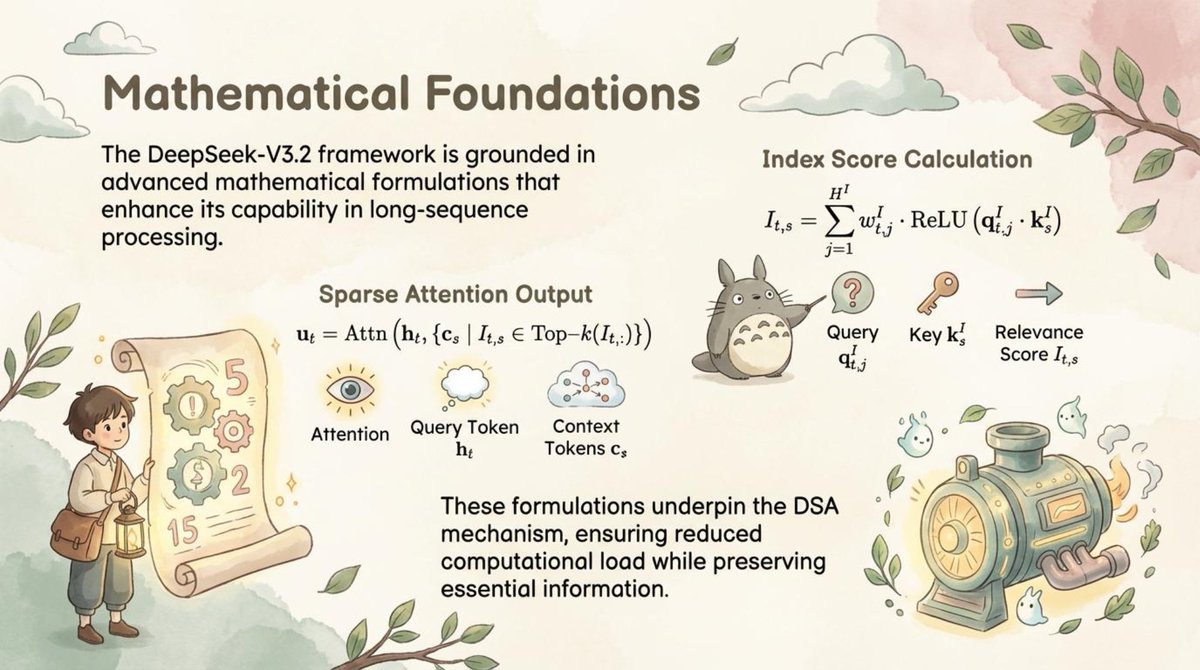
🚀 Paper2Slides is now open source! Transform research papers & technical reports into professional presentations with ONE click!
We've generated stunning presentation slides from the latest DeepSeek V3.2 paper in diverse styles - check them out and share your feedback!
🔥 Core Features:
- 📄 Multi-format support - PDF, Word, Excel, PowerPoint & more
- 🎯 Smart content understanding - Captures key insights, figures, formulas, tables & data points.
- 🎨 Custom styling - Professional themes with full personalization.
- ⚡ Lightning fast - High-quality PPT generation in minutes.
GitHub: https://t.co/zNxlFifDU3
Never build slides from scratch again! ✨ Come play with it ⭐!
#Paper2Slides #AIPPT
We just made an app that walks you through designing a novel protein with AI from scratch. Takes about 5 minutes, requires zero biology knowledge.
➡️ https://t.co/L3dg6H6BTU
The best part: we will actually synthesize 1000 of those protein designs in the lab and test their real world function as a therapeutic.