🧵1/
Delighted to announce our new study: Machine learning-based penetrance of genetic variants by @IainSForrest et al. published in @ScienceMagazine!
Paper: https://t.co/OLTEJI89PV
Press release: https://t.co/tp8aQrgMph
@AIHealthMtSinai@SinaiGenetics
David Reich is back.
He and collaborator Ali Akbari just published a paper that overturns a long-standing consensus about human evolution — that natural selection has been dormant in our species since the agricultural revolution.
By scaling ancient DNA sequencing and developing a new statistical method, they found that selection has actually sped up.
Selection went especially bonkers during the Bronze Age (around 3,000 years ago).
That's when gene frequencies for everything from immune function to body fat to intelligence were most in flux.
Over the last 10,000 years, selection pushed the genetic predictor of cognitive performance up by roughly a full standard deviation — most of it between 4,000 and 2,000 years ago.
After we finished recording, David sketched out on a whiteboard his new heretical model about who the Neanderthals really were. Luckily, I took out my iPhone and managed to record it.
He thinks the standard story (that Neanderthals are some separate archaic lineage we interbred with a little) just doesn't fit the evidence. Instead, he proposes that Neanderthals are essentially genetically-swamped modern humans.
A small population somewhere around the Caucasus invented Middle Stone Age technology roughly 300,000 years ago and expanded outward. The ones that moved into Europe interbred with local archaic humans, got genetically swamped, and became Neanderthals. The same expansion went into Africa, met much more diverged archaic Africans, and that mixture became us.
This means Neanderthals and modern humans share the same cultural ancestry — the only difference is which archaic humans they mixed with afterward.
David is a brilliant and rigorous scholar. It was a real delight to learn from him again.
0:00:00 – Ancient DNA suggests strong selection over last 10,000 years
0:16:24 – Natural selection intensified during the Bronze Age
0:35:40 – Why didn't evolution max out intelligence?
0:58:00 – Evolution is limited by time, not population size
1:09:40 – Why no farming before the Ice Age?
1:17:52 – The Neanderthal puzzle David can’t stop thinking about
1:54:40 – The methodology behind this breakthrough
Delighted to have our work on 🧬 resilience to 🩸cancer led by @GauravA_UK & amazing collaborators, including @KharasLab, published in @ScienceMagazine: https://t.co/lZDCy8CSBv 🧵
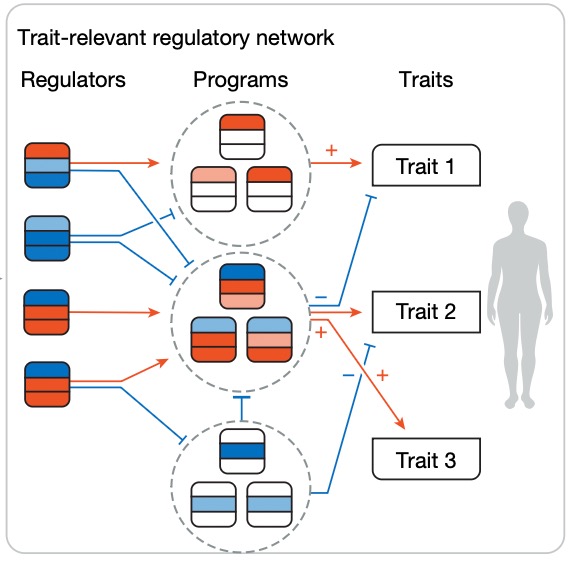
Modern GWAS can identify 1000s of significant hits but it can be hard to turn this into biological insight.
I'm excited to share our new work combining genetic associations and Perturb-seq to build interpretable causal graphs, out today in @Nature:
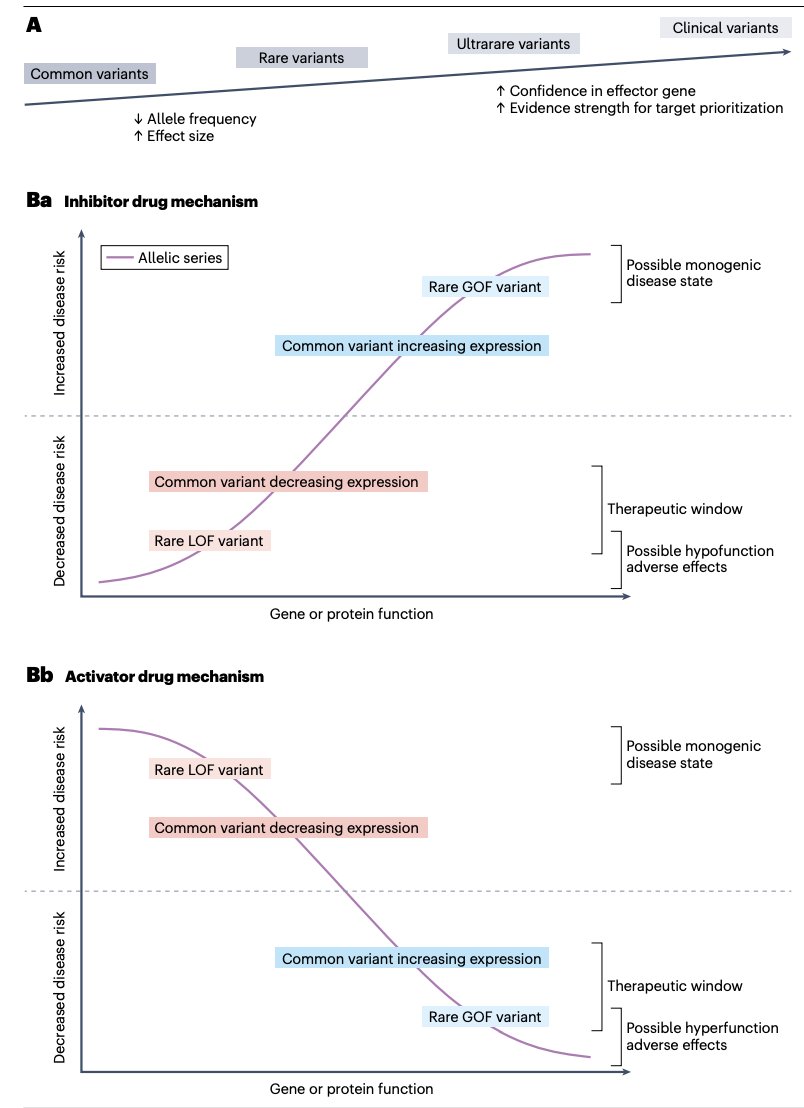
Successful drug development depends on identifying effective targets and optimizing drug design and clinical trial strategies. The authors review computational strategies, including ML, for integrating genetics into pharmaceutical pipelines https://t.co/aI7C1auULF
We are pleased to announce that our new study explaining the missing heritability of many phenotypes using WGS data from ~347,000 UK Biobank participants has just been published in @Nature. Please check out our manuscript here: https://t.co/PK2DIpshOQ.
Genetic analyses of eight complex diseases using predicted continuous representations of disease. A study by @DoGenetics@IcahnMountSinai & others @CellRepMethods#ASHG25 https://t.co/eeCpRW6uQI
Delighted to share our new research in @NatureComms.
@Aine_duffy94 et al. (2025): https://t.co/JSyc6hV1ho
“Human genetics can predict drug side effects.”
Our SE-GPS framework quantifies this risk and helps inform drug target safety from the start.
#Genomics#DrugDiscovery
🚨“Pathogenic” genetic variant does NOT = guaranteed disease 🚨
Yet, doctors and patients are still left to decide what to do with the label…
In our new @NatureGenet perspective, we explain why this is and how we can improve genetic risk interpretation🧵
Most genetic reports label variants as ✅ “pathogenic” or ❌ “benign.” But what patients really want to know is: what are my chances of getting the disease? 🚨 In our new Science study, we used AI + 1.3M patient records to better answer that…
Your DNA sequence reveals a rare mutation. Will you get the disease, i.e. penetrance? A new study using machine learning predicts this across 10 common diseases, 1,600 genomic variants @IcahnMountSinai@IcahnInstitute@ScienceMagazine https://t.co/FXU8PN2vgp
Insightful perspective on our ML penetrance paper on penetrance and variant consequences, in @ScienceMagazine nicely written by Harper Raiken and @_amelie_rocks
https://t.co/BfRoMl5KFh
11/
This approach is scalable and can be applied to many different types of variants and diseases.
It demonstrates how integrating ML + EHR + genomics can move us from categorical pathogenicity labels toward more data-driven and quantitative penetrance estimates.
/end
🧵1/
Delighted to announce our new study: Machine learning-based penetrance of genetic variants by @IainSForrest et al. published in @ScienceMagazine!
Paper: https://t.co/OLTEJI89PV
Press release: https://t.co/tp8aQrgMph
@AIHealthMtSinai@SinaiGenetics