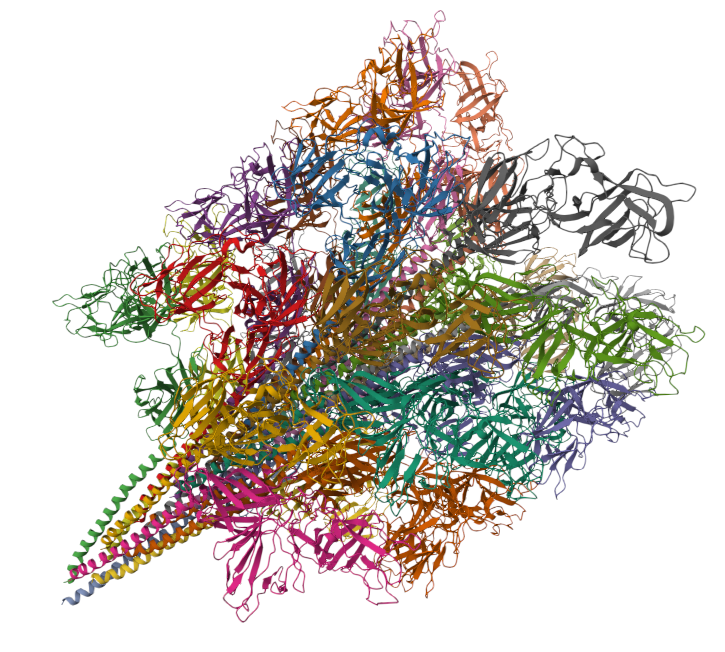
Characterizing AI-designed proteins requires quantitative biochemistry at massive scale. Enter Amplicon/Protein Bead Display (APB-Display), a fully in vitro platform that quantifies Kd's for >100,000 variants in <3 days (preprint link below!) @Stanford_ChEMH@czbiohub (1/n)
@ValerioCapraro I don't disagree with most of what you say in the paper. I think people routinely overestimate and misunderstand AI. But we need to build an epistemic framework for understanding *when* something is intelligent so when the problem arises in its true form we are prepared.
@ValerioCapraro I feel like this line of argument is not prepared for when- not if- they can do the things you're saying (rightfully, for now) that they cannot. When they have their own interior world, what then? Where do we draw the line?
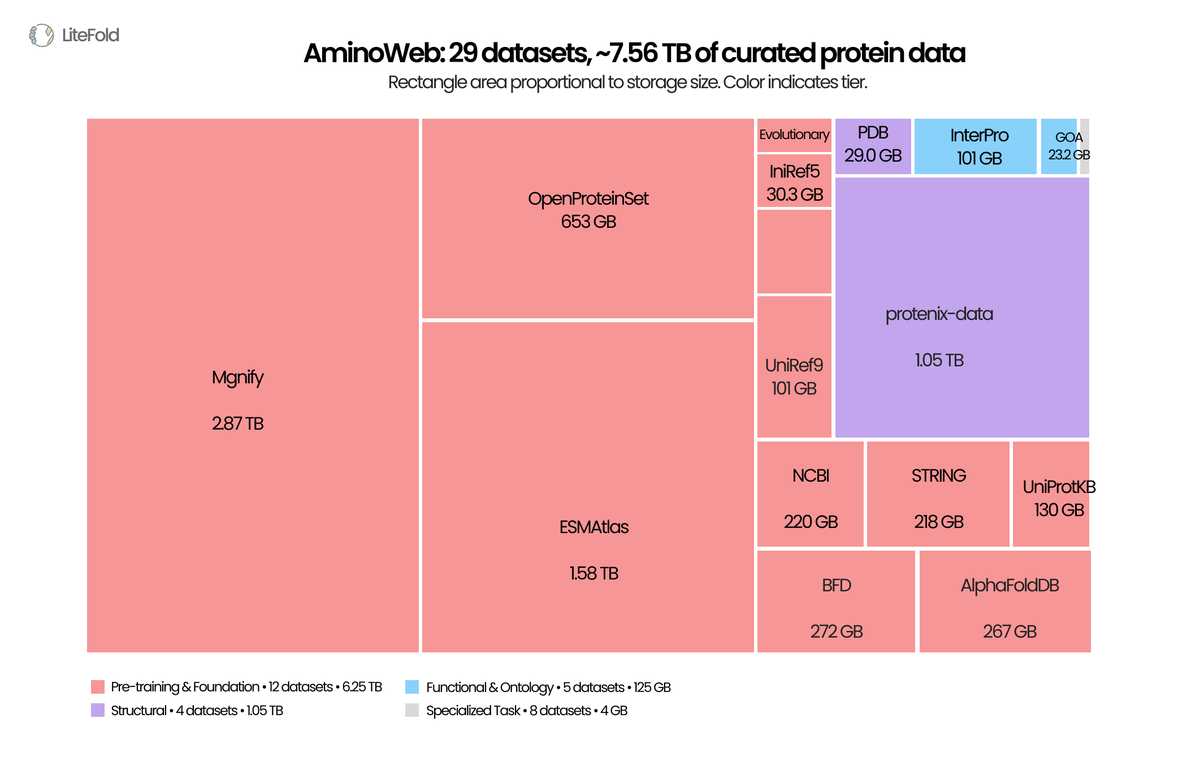
LLMs got FineWeb, The Pile, RedPajama, Dolma. Protein ML got per-paper supplementary tables and FTP mirrors scattered across a dozen institutions.
Today we're releasing AminoWeb on @huggingface : 29 cleaned, ML-ready protein datasets, ~7.5 TB total. Sequence, structure, function, MSA, variant-effect, stability, binding. UniProt, PDB, AlphaFoldDB, ESMAtlas, ProteinGym, MegaScale, Protenix, and more.
Typed Parquet. Homology-aware splits. Preserved score conventions. Full provenance per record.
Protein ML scaled architectures for years while the data layer stayed fragmented. We've also shared the full curation pipeline, case studies, and observations in the companion blog post.
Access the data: https://t.co/elQ7pzpNkG
Read the release blogpost: https://t.co/28yFU2m9Jc
Today we're announcing ESMFold2, an open scientific engine to power prediction, design, and discovery across protein biology.
The new model delivers state of the art performance on protein interactions, especially antibodies, a critical modality for therapeutics.
We have designed and validated miniprotein binders and single chain antibodies across five therapeutic targets that are important in cancer and immunology. We are seeing very high success rates, and affinities at levels consistent with therapeutic activity.
We’re also releasing an atlas of 6.8 billion proteins, and 1.1 billion predicted structures.
ESMFold2 is built on a state of the art language model that has been trained on billions of protein sequences.
A world model of protein biology emerges through language modeling.
We’ve used the techniques of mechanistic interpretability developed to understand large language models to understand the concepts ESM uses to represent proteins.
The model’s representation space has a compositional organization of features across scales, levels of complexity, and abstraction, that reflects and mirrors the understanding of protein biology developed through a century of empirical science.
This understanding emerges without prior knowledge, just from language modeling of protein sequences.
Language models are becoming a powerful substrate to understand and program biology.
The design of protein interactions is one of the most fundamental problems in biophysics, and has critical implications for the discovery of new medicines. A simple gradient based search with the model was able to discover high-affinity protein binders.
I'm excited by the potential this has to accelerate basic science and the understanding of proteins. And especially for the new avenues it opens up for therapeutic design and medicine.
Sandpiper 2 is up. 913,000 metagenomic community profiles w GTDB R232 via SingleM, 200k more than 1.0. https://t.co/YYxXe1l8Cz
GlobDB coming.
Thanks to @aroney_samuel@IAmTotesBrett Josh Mitchell and especially the new kid @StefanHerh
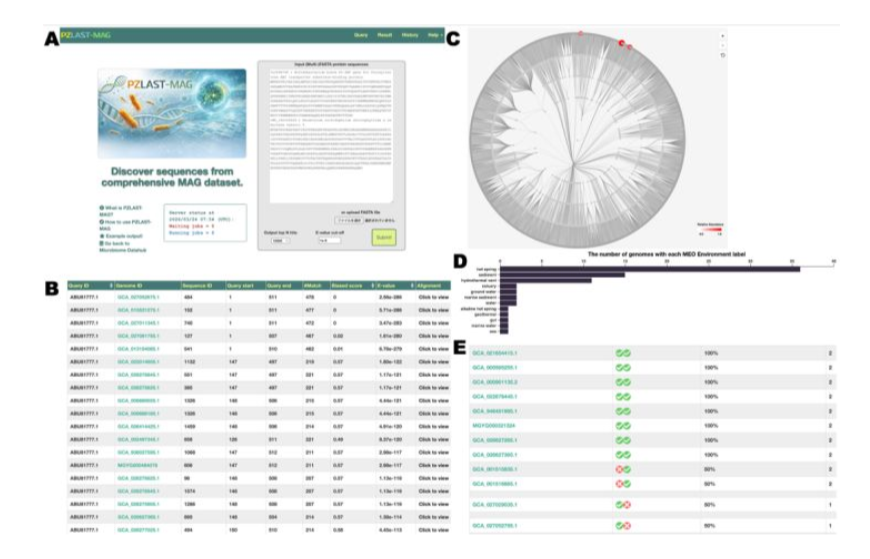
🔍 New in Bioinformatics Advances: "PZLAST-MAG: Full length protein sequence similarity search server of large-scale MAG proteins"
Read it here: https://t.co/8zBMHZFqth
so fun to see people on here contorting their thought processes to justify their belief that frontier AIs are static tools without emergent capabilities
We wrote a review on wet-lab validated deep learning protein design methods.
I was very impressed that @kosonocky was able to keep all these papers straight and also fit this into the world limits!
I’m looking for an automated way to read others’s scientific data without giving credit or acknowledgement, and also claim full credit for insights from it. And I want it to have a fitting name
OAI: say no more