Decoding aging-dependent regenerative decline across tissues at single-cell resolution
https://t.co/09rLDIWZWs
Open stem cell research from Cell Stem Cell.
happy to see our cell2fate model out at @naturemethods. It is a completely new take on RNA velocity to disentangle complex cell trajectories from scRNA data & we have some very cool applications coming up! https://t.co/PpOJUvradp
1/In his excellent article WSJ columnist @david_wainer lays out the massive change which is happening in the American BioTech ecosystem & the threat that rising Chinese Biotech companies pose in almost the same way that DeepSeek has done in AI. The 🇨🇳 impact is huge! $XBI 🧵👇
Now out in @ScienceMagazine we present 'Genome-shuffle-seq': a method to shuffle mammalian genomes and characterize the impact of structural variants (SVs) with single-cell resolution in one experiment.
https://t.co/dKnuHkQ0W9
Excited to share our latest paper from a collab w Patrick Sullivan and others where results from two different data-driven approaches (GWAS/human-snRNAseq and fMRI) converge on the amygdala and hippocampus as key regions for schizophrenia: https://t.co/R1Zd32vMmX 🧵
Super excited to finally share our new manuscript, out today in @NatureCellBio! This is the culmination of 6 years of work examining a seemingly simple question: What are the metabolic requirements for overcoming mitochondrial stress?
https://t.co/btsItt3xog
We are thrilled to share our new single-cell foundation model, Tabula (preprint: https://t.co/hxGu7X8p4z; package: https://t.co/auxfUPZwhS)—a privacy-preserving predictive foundation model for single-cell transcriptomics, leveraging federated learning and tabular modeling.
Over the past year, we’ve seen a surge in foundation models for single-cell genomics, where genes are often arbitrarily ordered to mimic NLP paradigms. Furthermore, as we start to train on large datasets comprising thousands of individuals, the ethical and privacy concerns arise as well. To address these challenges, we introduce Tabula: a federated-learning-based, privacy-preserving foundation model that explicitly represents single-cell data using tabular modeling. Tabula demonstrates excellent performance across diverse tasks, including cell type annotation, multi-omics and multi-batch integration, gene imputation, denoising, and both gene perturbation and reverse perturbation predictions. Very excitingly, as one of the first examples of a truly predictive foundation model, Tabula accurately uncovers pairwise and even combinatorial regulatory logic across diverse biological systems, including hematopoiesis, pancreatic endogenesis, neurogenesis, and cardiogenesis, all of which have very well validated regulatory networks. For more details, please see @JiayuanDing 's excellent post here: https://t.co/db1qeUgic5
This is really an amazing collaboration with three brilliant young trainees, including @JiayuanDing Jianhui (@JilinJJ) and Shiyu (@shiyu_jiang23) and two other labs Jiliang (@tangjiliang), and Min Li..
Kudos to @JiayuanDing , the incredibly talented PhD student in my lab who led this project! Jianyuan is currently on the faculty job market and would be an outstanding addition to any institution. Please consider him and feel free to reach out to Jianyuan or me for more information.
Additionally, Shiyun @shiyu_jiang23 , who contributed to innovative approaches for pairwise and combinatorial perturbation prediction, is applying to PhD programs. Please consider this rising star to join your PhD program as well!
This work represents another important advance in my lab’s long time vision to establish a predictive “virtual embryo” model for human health. We are currently extending this approach to 3D Spatial Transcriptomics data as we previously reported in Spateo (https://t.co/fSexZQeejN) . If you are a developmental biologist, technology developer, or a machine learning expert, please consider joining us on this exciting journey, please reach out regarding potential positions at all levels in my lab (https://t.co/qpOqV6MRvg. Email: [email protected]). We also highly welcome graduate students from Stanford
@StanfordEng@DevBioStanford@ChemSysBio@StanfordData@StanfordAILab
for rotations in my new lab. I am excited about many collaboration opportunities within Stanford and the broader Bay Area as well.
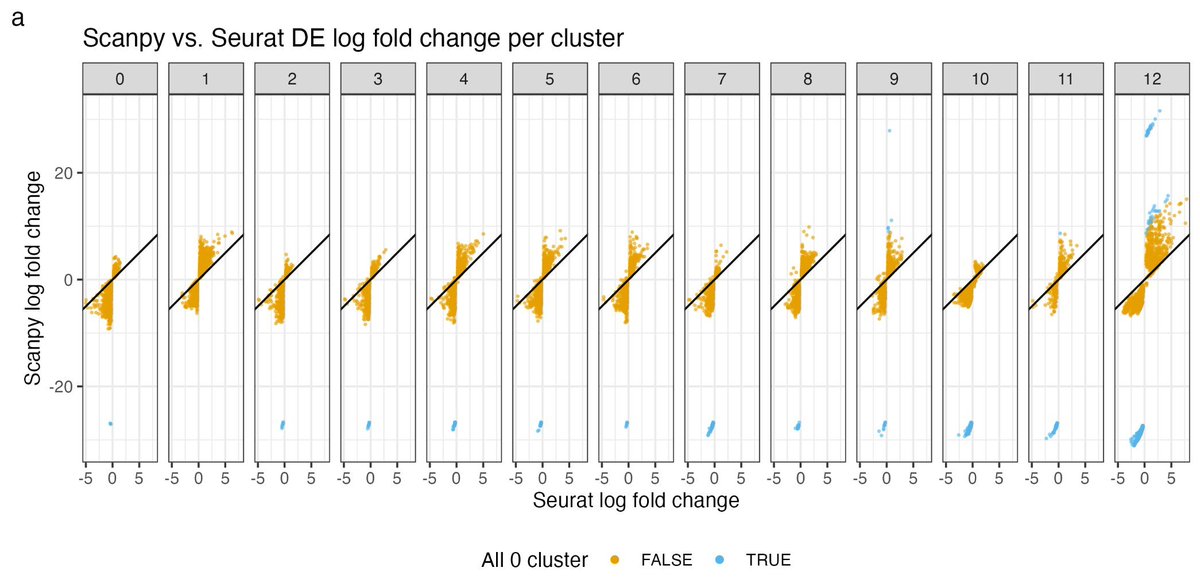
chatomics! Scanpy and Seurat marker gene log2Fold change has a big discrepancy!
Do you understand log2Fold change in single-cell RNAseq data? https://t.co/v7T1W24R8C
A single-cell atlas of aging tissue demonstrates shared multi-omics of aging and cancer hallmarks
https://t.co/DgBcpvYegc @NatureAging@jacksonlab @justsaysinmice @OlgaAnczukow
Our paper on single-cell splicing QTL is now published as a cover article in Nature Genetics. https://t.co/jHas1xKi01
Congratulations to co-first authors @chitian51616, @Yihan720, and Yuntian Zhang! Huge thanks to AIDA leaders @shyam_lab , Woong-Yang, @JayShin and Kian Hong!
In collaboration with Reuben Saunders, @JswLab, and Xiaowei Zhuang, we are very excited to release Perturb-Multi: a platform for pooled multimodal genetic screens in intact mammalian tissue.
Check it out!
https://t.co/iJ8hi3ddz4
Exploring public cancer gene expression signatures across bulk, single-cell and spatial transcriptomics data with signifinder Bioconductor package https://t.co/rrLwynvji8
Leveraging @10XGenomics Flex technology, we develop fixed-cell Perturb-Seq with direct hybridization to sgRNAs. The ability to use fixed cells makes large-scale in vivo Perturb-seq drastically easier and cheaper, while accurately measuring cellular gene expression.