Official Twitter for the Gómez-Bombarelli group @MIT_DMSE | We use atomistic simulations and ML for accelerated materials design | Managed by group members
Thrilled to announce the final preprint of my PhD!
We introduce PackFlow, a flow matching method for generative molecular crystal structure prediction, and post-trained via reinforcement learning on MLIP energies and forces.
Paper: https://t.co/wFbxV7RQb4
@RGBLabMIT
We'll be giving a public talk about this work tomorrow, January 28, at 11:30 AM ET (8:30 AM PT, 4:30 PM GMT)! It'll be hosted by LeMaterial, an initiative from @entalpic_ai and @huggingface. We'd love to see you there!
🔗https://t.co/Htj360egSZ
📅 https://t.co/38bmrkCM28
Scientific foundation models are converging to a universal representation of matter.
Come chat with us at #NeurIPS! We (@SoojungYang2@RGBLabMIT) have an oral spotlight at the #NeurIPS#UniReps workshop and will also poster at #AI4Mat.
🖇️: https://t.co/H8ZRlpvJUj
🧵(1/5)
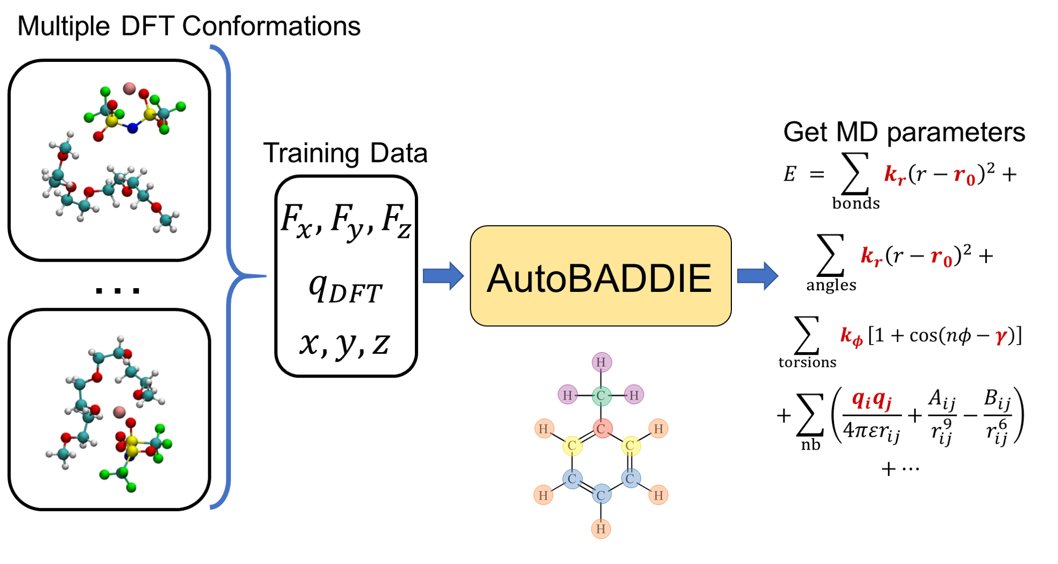
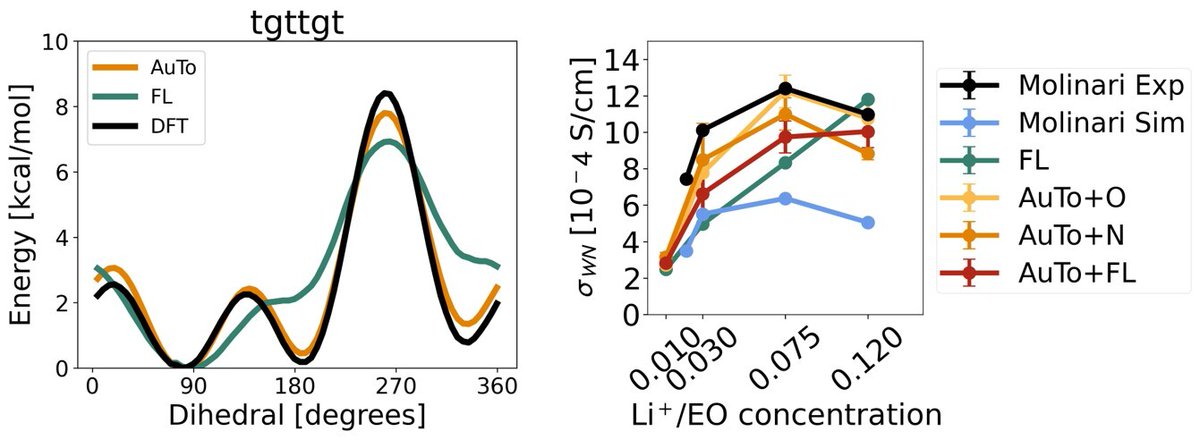
Our work on "End-To-End Learning of Classical Interatomic Potentials for Benchmarking Anion Polarization Effects in Lithium Polymer Electrolytes" is out now in Chemistry of Materials! https://t.co/JSDwQwv8VZ
We also find that previously-parameterized classical potentials model two separate anion polarization states that drastically influence resulting lithium solvation and transference.
The surprising ineffectiveness of molecular dynamics coordinates for predicting bioactivity with machine learning
1. The study challenges the assumption that molecular dynamics (MD)-derived coordinates are superior for machine learning-based bioactivity predictions, revealing that they often underperform compared to minimum-energy conformations.
2. Using over 2600 protein-ligand complexes, the authors systematically compared MD-derived and minimum-energy coordinates, employing three descriptor sets and machine learning algorithms like Random Forest, XGBoost, and Support Vector Regression.
3. Surprisingly, MD-derived conformations failed to consistently outperform minimum-energy structures, even though they provide dynamic representations of molecular interactions.
4. In certain cases, ensemble averaging of MD-generated snapshots improved predictive performance slightly, but the benefits were not proportional to the computational costs.
5. Extended Connectivity Fingerprints (ECFPs), a 2D molecular representation, outperformed MD-based models in many cases, questioning the utility of complex 3D data for predicting bioactivity.
6. The findings highlight a critical need for better 3D and dynamic molecular representations. The study suggests exploring geometric deep learning or incorporating protein information to improve machine learning models.
7. The authors propose a tiered approach: using fast, simpler methods like ECFPs for initial screening, followed by MD-based predictions for refining top candidates.
8. The study serves as a wake-up call for molecular machine learning, emphasizing the importance of balancing data complexity, computational cost, and predictive accuracy.
@fra_grisoni@DerekvTilborg@Rza_ozcelik
📜Paper: https://t.co/6b6GxztgQ9
#MachineLearning #DrugDiscovery #MolecularDynamics #Bioinformatics #AI
📢New preprint out! We constrain the molecular generation space to follow the "symmetry" of patented molecules that are likely to be synthesizable. Achieved with "symmetry-aware" fragment decomposition, and a constrained Monte Carlo Tree Search generator. https://t.co/NWidW2Wx9y
Zero-shot extrapolation for out-of-distribution (OOD) chemical property prediction is an important step towards high-performance materials discovery. Check out our spotlight at the #NeurIPS AI for Accelerated Materials Design Workshop! https://t.co/wHxezk4zD7
I will always upvote bogus enthalpy-entropy compensation.
My thesis advisor, J Casado, loved this paper. I remember doing the calculations in undergrad kinetics class some 20 years ago
Instead of the typical (somewhat technical) lecture, we discussed a couple of papers, including a favorite J. Chem. Ed. activity of mine: "Chemistry from Telephone Numbers: The False Isokinetic Relationship." It's a lot of fun. Do check it out. 2/3
https://t.co/uvFEdlESXK
MERGED NETS 💎📄
I can’t describe 350+ new nets in 280 signs, so just watch the animation & read our new paper in @ScienceMagazine 👨🎨💎👨🔬 with @Eddaoudi_FMD3.
In short, we merge nets together and get new topologies perfect for designing mix-ligand #MOFs.
https://t.co/xnARltj9dO
Applying to DMSE? The DMSE Application Assistance Program (DAAP) offers support for students from underrepresented groups in science and engineering. You’ll be paired with a grad student mentor to guide you through the application process. Apply by Nov 1. https://t.co/4H3Eq1gHxz
In tomorrow’s Wulff Lecture, DMSE’s Professor Antoine Allanore will explore greener iron and steel production processes, highlighting innovations that use electricity instead of carbon. October 23 | 4 pm | 6-120 https://t.co/Ngj6F32juC
Code💻: https://t.co/o2VV1pjJMV
HuggingFace paper page 📰: https://t.co/55fsBHOAgu
Shout out again to my amazing collaborators 🙌🙌🙌 and @thjashin for insightful discussion ❤️❤️❤️! (11/11)
Discrete generative models use denoisers for generation, but they can slip up. What if generation *isn’t only* about denoising?🤔
Introducing DDPD: Discrete Diffusion with Planned Denoising🤗🧵(1/11)
w/ @junonam_@AndrewC_ML@HannesStaerk@xuyilun2 Tommi Jaakkola @RGBLabMIT
Final group photo of the @cecamEvents workshop on "Advances in catalytic reactivity simulations under operando conditions"!
Thanks to all participants for your valuable contributions and stimulating discussions
and to @IITalk@fondazione_fair for support & CASALE SA for sponsor
Please help repost and spread the word. 🙏 My research group at @UBuffalo's Materials Design & Innovation (@UBengineering & @UBCAS) is recruiting multiple graduate students! https://t.co/IPnthYoS1F
If you are interested in combining 🤖 data science and machine learning with ⚛️ materials physics and surface chemistry to design materials and interfaces for⚡ decarbonization and 🧪 sustainability, please visit our website and reach out to me by email (see the link above for more details).
#sciencetwitter #research #chemistry #materials #machinelearning #artificalintelligence