@_JasonDerks is presenting at iSCMS! Come by this Saturday afternoon around 2pm to hear his talk: "Increasing proteomics throughput by multiplexing in the mass and time domains".
https://t.co/IY8ztiXV1H
Vimentin in Fibroblast cells is likely functionally analogous to Krt5 in Basal cells.
It’s amazing that single cell proteomics can enable these types of finding. Check out the work from @_AndrewLeduc for more details!
The big one is finally out!! In this paper, we set out to provide insight into the fundamental question;
How do the individual cells from complex tissues regulate their proteomes?
Brief summary of our findings 👇
https://t.co/4dRXpGJTGn
I was super excited to learn from @NikoMcCarty's essay, "How to Scale Proteomics", that there's an FRO working on high-throughput single-cell proteomics (@ParallelSqTech), and even more excited to see the progress they've made in just 2 years via their preprints.
So here's a video about it!
And some links you should check out⬇️
PTI will be represented at #ASMS2025 with with two posters (TP203 and ThP 533) and two talks (TOA 2:30 pm and WOA 8:30 am). Find us to hear about our PSMtag, and applications in Alzheimer's and aging biology!
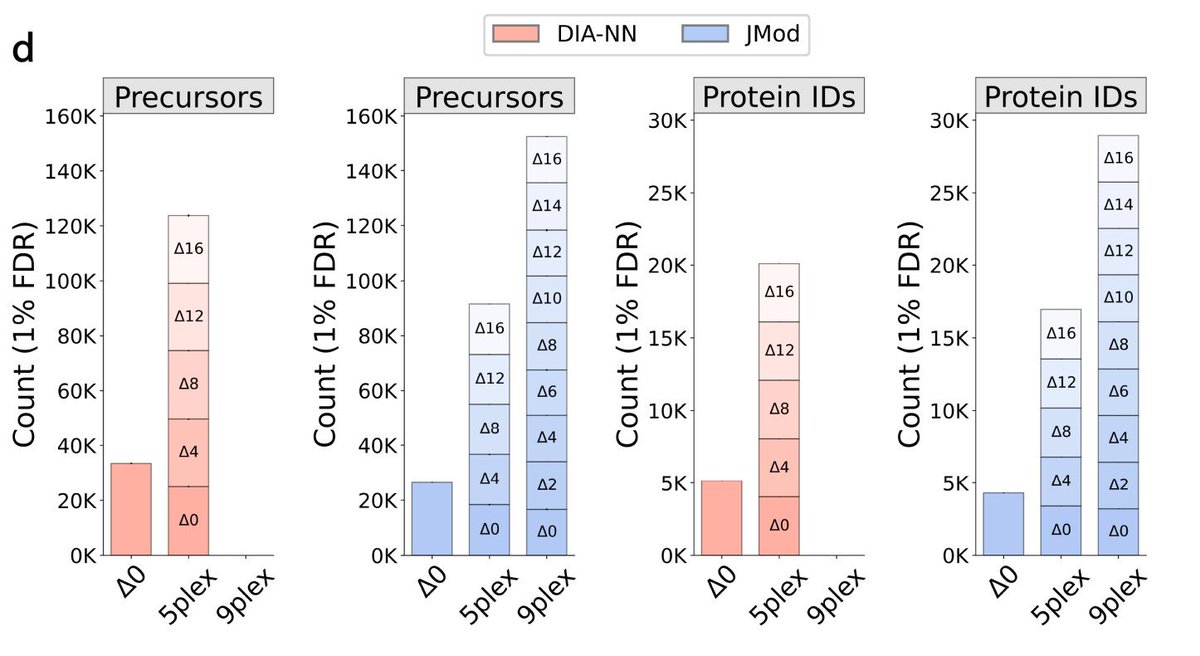
We just released JMod: an open source software supporting multiplexed DIA.
It supports parallelization in both the mass domain (plexDIA) using small mass offsets and in the time domain (timePlex).
It enabled 27-plex DIA & much further scaling 🚀
https://t.co/u1I1QAZxSN
Links to our preprints:
timePlex (multiplexing with time-offsets): https://t.co/tDeYgJuLei
JMod (search software for joint-modeling): https://t.co/HBOrxeiUGf
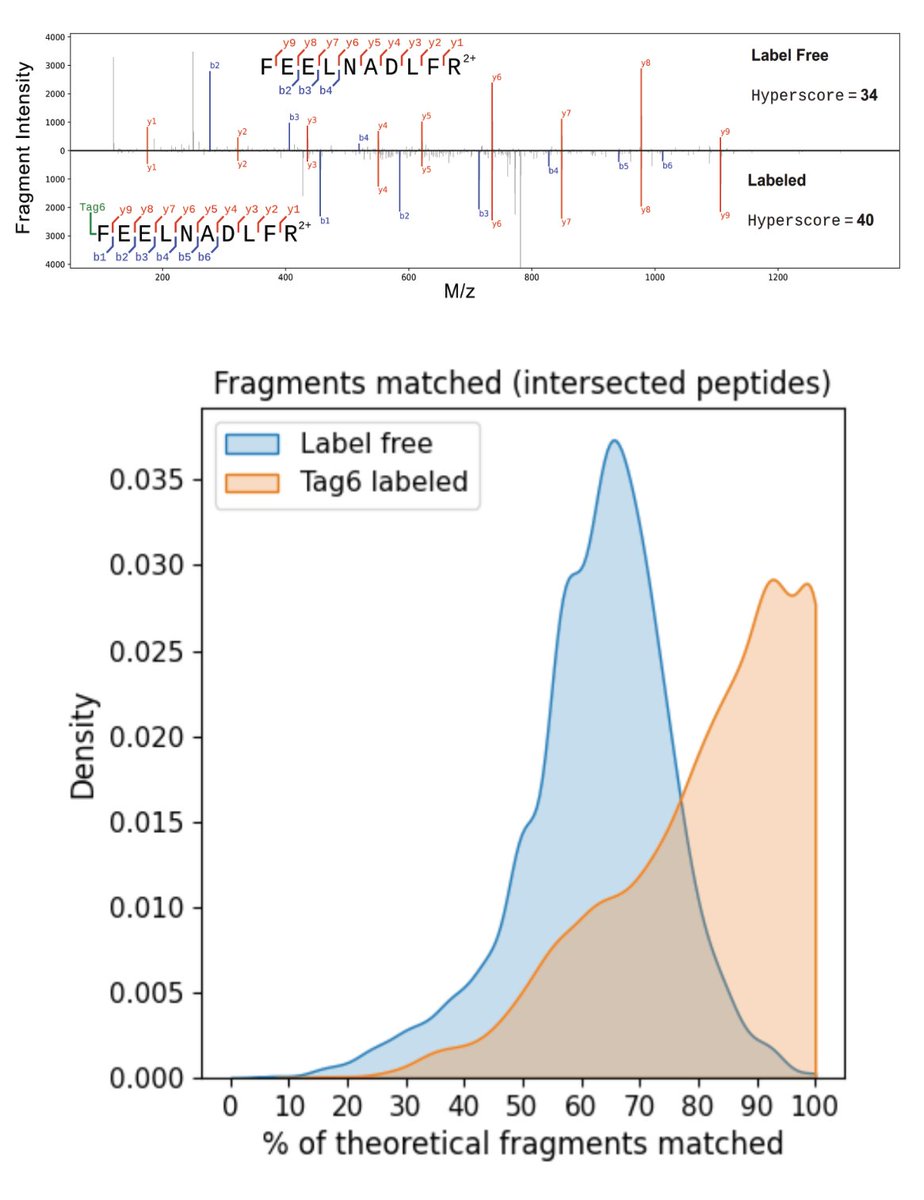
PSMtag (novel mass tag for 9-plexDIA): https://t.co/ei449lDkpr
We are excited to introduce ‘time’ as a new domain for proteomics multiplexing!
It enables:
-Label-free multiplexing
-Combinatorial multiplexing with plexDIA
Using 9-plexDIA and 3-timePlex together we demonstrate 27-plex DIA 🚀
https://t.co/tDeYgJuLei
This work was a collective effort by members of @ParallelSqTech, enabled analytically through the development of specialized search software (JMod) and through mass-tag engineering (PSMtag).
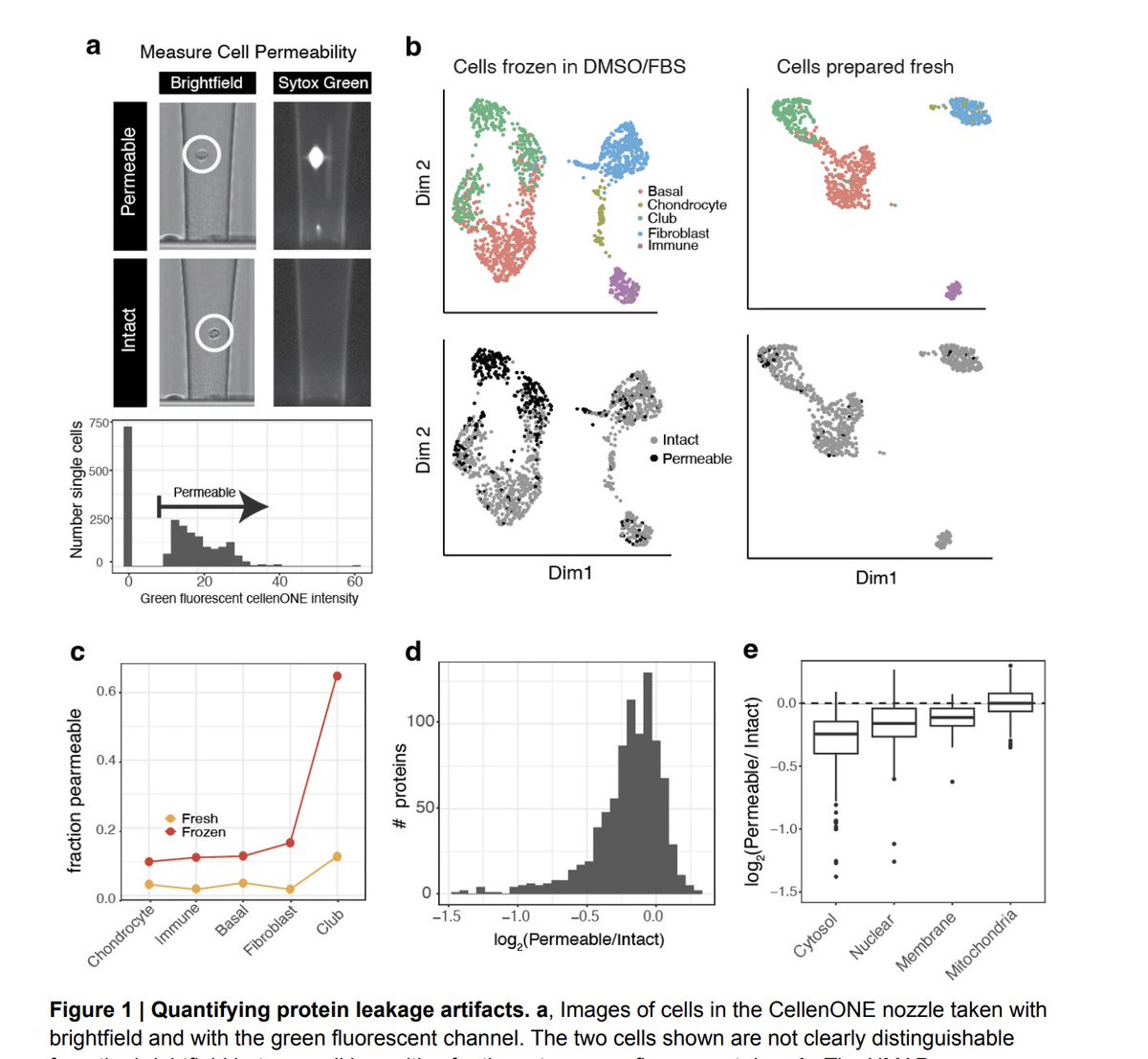
Our paper on the impact of protein leakage due to loss of membrane integrity for single cell proteomic sample prep is out in @NatureComms!
We analyze thousands of primary single cells from murine tracheal tissue and record membrane permeability🧵
https://t.co/uj2Wpz8LRs
Protein samples can be multiplexed to increase throughput.
In 2023, @ParallelSqTech set out to make tags (barcodes) that not only increase throughput but also enhance peptide sequence identifications: https://t.co/RsZzKeWihh
We will share results soon: https://t.co/rJQ6PSkcmI
We have released DIA-NN 2.1.0, download link https://t.co/4x5BsyVfiC, release notes https://t.co/yTctBkcoVx.
It is a minor update, main highlights are:
- Built-in support for Thermo .raw (Windows + Linux)
- New QC metrics in PDF report & visualising trends across the experiment
We report many proteins not predicted by the genetic code.
They are stable & abundant O( 10³ ) copies / cell.
Generative mechanisms include codon-anticodon mismatches & RNA modifications.
Their abundance depends on codon frequency & protein stability.
https://t.co/lAxvulC4zx
Check out our work on identifying and providing solutions for protein leakage artifacts associated with sample preparation.
It all started out with our struggle to explain a strange bi-clustering pattern in our data.
A new preprint:
While analyzing proteomes of cells from mammalian tissues, @_AndrewLeduc identified protein leakage from permeabilized cells.
We identified clear patterns that can be used to detect & mitigate this issue:
https://t.co/CSopooo47J