Enhanced Diffusion Sampling: We develop a framework for efficient rare event sampling and free energy calculation with diffusion models. We introduce Metadynamics and Umbrella Sampling for diffusion models.
@MSFTResearch#MachineLearning#MD#Biology
https://t.co/NC6QNrCP85
ParametrizANI found a home!
Our paper is now published in JCIM!
Making ML-based force field parametrization open and easy for everyone.
With TorchANI, @RDKit_org & @openmm
🔗 https://t.co/6SrjSMkF13
@JCIM_JCTC@palermo_lab@sou_svk
Studenti Medicinskog fakulteta u Beogradu, zajedno sa profesorima, objavili su rad u jednom od najprestižnijih svetskih medicinskih časopisa, BMJ (British Medical Journal).
Rad ukazuje na to da zdravlje ne može da napreduje u okviru represivnih režima; njegova zaštita zavisi od slobode, solidarnosti i zajedničke odgovornosti.
Čestitamo, kolege! ���
https://t.co/NYciLOrzqp
🎲 Our paper on the genetics, energetics, and allostery in proteins with randomized cores and surfaces is out today @ScienceMagazine!
🧬 By charting a protein’s sequence universe, we could rationalize which versions were kept through evolution �� and why many stable ones were not.
Today in the journal Science: BioEmu from Microsoft Research AI for Science. This generative deep learning method emulates protein equilibrium ensembles – key for understanding protein function at scale. https://t.co/WwKjj5B0eb
Mapping the off-target effects of every FDA-approved drug in existence (EvE Bio)
6.2k words, 29 minutes reading time
https://t.co/3tjPmzwRZw
this is a very long essay over @EvEBiotech, a data collection effort that you should know about (it's associated w/ @Convergent_FROs!)
Never stop being a proud physicist @DimaKrotov , it's a pleasure working with you 🥳.
https://t.co/U5gimfI6B8
"Computation is a physical process. We can study the flow of bits just as we study the flow of atoms"
More juicy Dima snippets in 🧵
Excited to unveil Boltz-2, our new model capable not only of predicting structures but also binding affinities! Boltz-2 is the first AI model to approach the performance of FEP simulations while being more than 1000x faster! All open-sourced under MIT license! A thread… 🤗🚀
Residue conservation and solvent accessibility are (almost) all you need for predicting mutational effects in proteins
1.A simple model called RSALOR achieves performance on par with or better than most state-of-the-art tools in predicting the effects of mutations on proteins — using only two features: sequence conservation and solvent accessibility.
2.RSALOR is based on a log-odds ratio (LOR) of wild-type vs mutant residue frequencies from an MSA, scaled by the relative solvent accessibility (RSA) of the residue. No training, no neural networks, no black boxes.
3.The model is unsupervised, highly interpretable, and fast — it evaluated 2.5 million mutations from the ProteinGym benchmark in under 20 minutes on a standard laptop with 8 CPUs.
4.Despite its simplicity, RSALOR outperforms or matches 27 top predictors — including 19 protein language model (pLM)-based methods — across multiple mutation effect categories: stability, fitness, binding, expression, and activity.
5.In single-mutation prediction tasks, RSALOR achieves an average Spearman correlation of 0.473 across all ProteinGym DMS datasets, second only to ProSST, a much more complex structure+pLM hybrid.
6.The model nearly preserves symmetry: a mutation from A→B has nearly the opposite effect of B→A. This is a desirable feature often missing in other methods, and important for protein stability prediction.
7.RSALOR makes use of high-quality MSAs and AlphaFold structures. The predictions are robust to the use of homologous or non-exact structures, due to RSA's insensitivity to small changes in conformation.
8.For multiple mutations, RSALOR assumes additivity (i.e., no epistasis). Even so, it performs surprisingly well, showing a 0.484 Spearman correlation across all mutations in ProteinGym.
9.RSA contributes differently depending on the task: it significantly boosts performance for stability and binding predictions, but offers less improvement for fitness, activity, or expression.
10.Notably, adding RSA to other models (via the RSALOR formulation) improves their performance too — even for predictors that already use structural inputs. RSA remains an underutilized but powerful feature.
11.RSALOR is available as a Python package on PyPI and GitHub. No model fitting required, no external dependencies, and minimal compute — making it ideal for practical, large-scale protein design or analysis.
12.This work questions the need for highly complex models when simple, biologically motivated features — conservation and accessibility — can be equally effective. It also highlights where current deep models may still fall short.
💻Code: https://t.co/rBlvh5a9vD
📜Paper: https://t.co/QiNUoCwasS
#ProteinDesign #MutationalEffects #Bioinformatics #ComputationalBiology #MachineLearning #ProteinEngineering
Thrilled to share our new paper where we introduce a multiplexed hydrogen–deuterium exchange MS (mHDX‑MS) method that can measure hundreds of protein domains’ conformational energy landscapes—all in a single experiment!
https://t.co/E6QfRWiDVf
The code & camera-ready version of our #ICLR2025 paper on "Multi-domain Distribution Learning for De Novo Drug Design" are now available
📚 Paper: https://t.co/NCeHVltqm8
💻 Code: https://t.co/0GGMiOODla
(1/4)
DeepSeek-R1 is great, but it still does not handle genomics yet :)
🚀 We’re thrilled to announce that IntegrAO (pronounced "integral") 🧬🔬—our new multi-omics integration framework—has been published in Nature Machine Intelligence!
📑 Paper: https://t.co/KziOfks2iL
💻 Code: https://t.co/pvgyIVuDF0
🔬 What is IntegrAO?
IntegrAO (Integrate Any Omics) is an unsupervised platform designed to tackle the challenges of incomplete multi-omics data. Crucially, it can be seamlessly transformed into a prediction model after integration, enabling robust classification of new patient samples—even when only partial omics data is available.
🧠 Why IntegrAO?
Effective cancer stratification depends on comprehensive multiomics data, yet real-world clinical datasets are often incomplete. Accurately assigning new patients to predefined subtypes, even when only partial omics are available, is crucial for personalized care but remains a major barrier to the clinical adoption of multiomics. Existing methods struggle with missing data and rarely address the essential task of classifying patients with partial profiles. IntegrAO excels at overcoming these challenges.
🌟 IntegrAO Highlights:
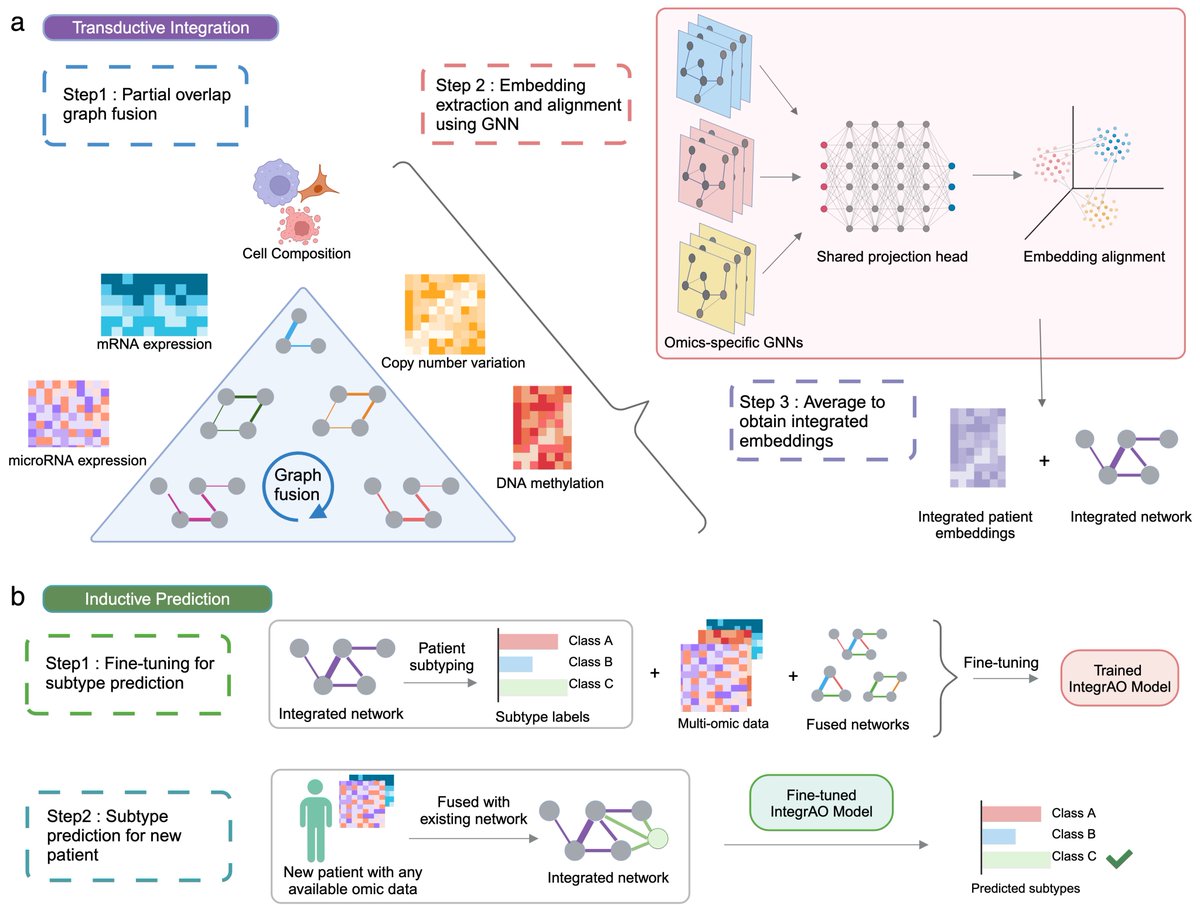
- Transductive Integration: Integrates patient graphs with partial overlaps across any omics sources. Utilizes GNNs to produce unified patient embeddings, preserving critical biological and clinical signals.
- Resilience to Missing Data: Demonstrates robust performance on heterogeneous, incomplete datasets from multiple cancer cohorts. Maintains accuracy despite varying degrees of missing omics.
- Inductive Prediction: Converts into a prediction model via supervised fine-tuning. Accurately classifying new samples with partial data into existing subtypes is crucial for precision medicine.
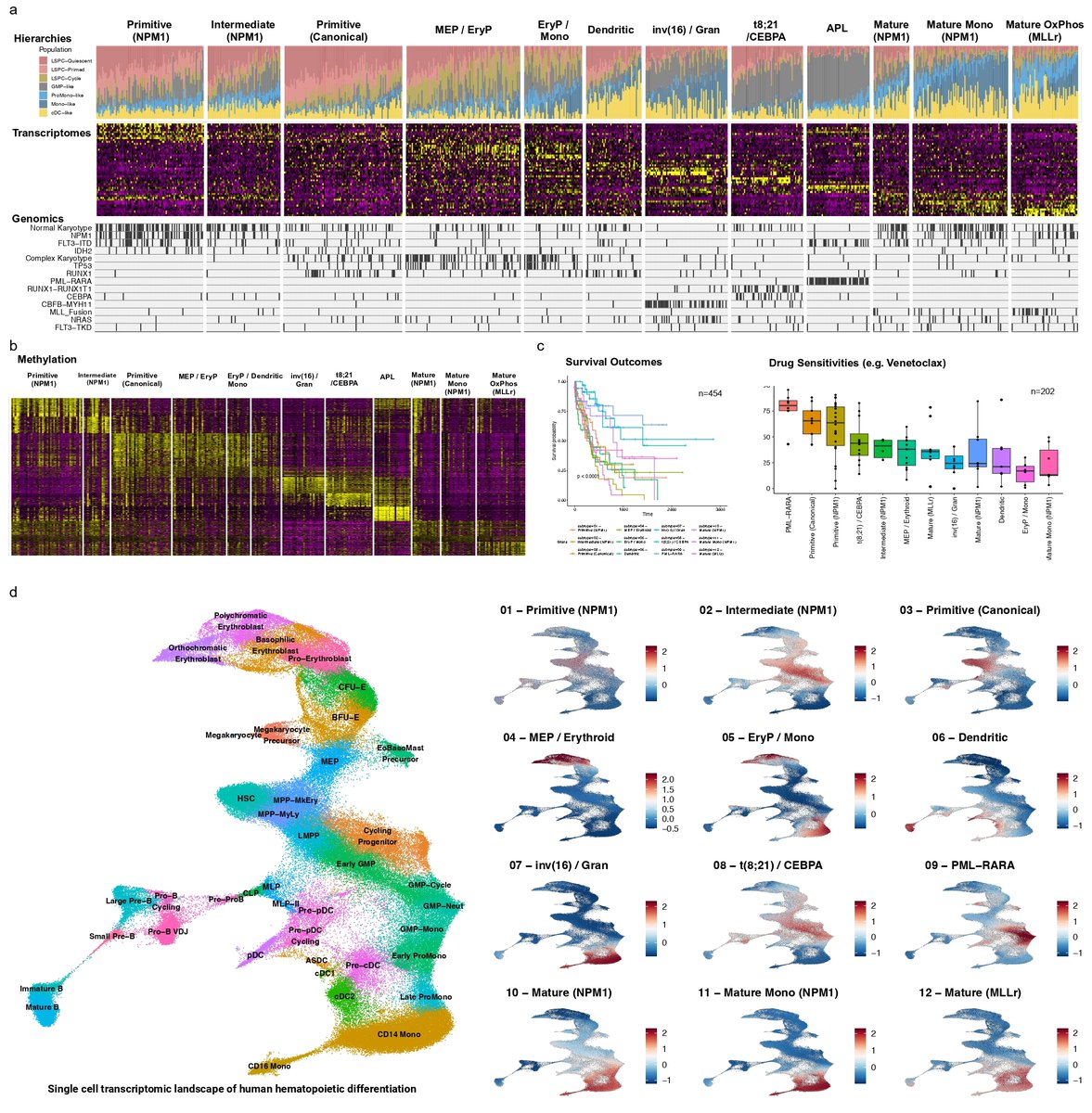
- Real-World Impact: Comprehensive case study on acute myeloid leukemia, revealing clinically and biologically relevant subgroups.
Shoutout to Rex Ma (@RexMa9), an incredible PhD student in the lab, for his exceptional leadership on this project! Huge thanks as well to our amazing collaborators: Andy G. X. Zeng, Benjamin Haibe-Kains, Anna Goldenberg, and John E. Dick! 👏🎉
@VectorInst@UHN_Research@UofTCompSci@UofT_TCAIREM
Long-range ML potentials strike again! 🚀 We benchmarked LES on diverse systems—molecules, solutions, and interfaces. Learning just from energy & forces, LES gives the most accurate potential energy surfaces, and physical charges, dipoles, quadrupoles!
https://t.co/73zx6popZW
🚨 Excited to announce the release of the DL4Proteins notebook series! 🌟
Learn AI for protein design with hands-on Colab tutorials inspired by the groundbreaking work of 2024 Chemistry Nobel Laureates David Baker, Demis Hassabis, & John Jumper.
https://t.co/pi1tEwr0ij
#HighlightOfTheWeek Cosolvent molecular dynamics are an increasingly popular form of MD simulations where small molecule cosolvents are added to the system, helping to identify cryptic and allosteric pockets in proteins.
#compchem
https://t.co/XGn48GelmP
ProtSCAPE: Mapping the landscape of protein conformations in molecular dynamics
1. ProtSCAPE is a deep learning architecture designed to map protein conformations from molecular dynamics (MD) simulations, using a novel combination of learnable geometric scattering with dual attention mechanisms.
2. The model employs geometric scattering to capture both local and global protein structures, representing proteins as graphs, which are then processed by a transformer with dual attention—focusing on residues and amino acids.
3. ProtSCAPE’s latent representations are temporally coherent, allowing it to capture conformational transitions, such as phase changes between open and closed states, and stochastic switching between meta-stable conformations.
4. Unlike conventional MD trajectory analysis that often misses complex transitions, ProtSCAPE excels in generating detailed low-dimensional representations that retain structural and temporal context, enabling enhanced visualization and downstream analysis of protein dynamics.
5. ProtSCAPE effectively generalizes from short to long trajectories and from wild-type to mutant proteins, offering insights into how mutations can affect the protein conformational landscape.
6. The model can interpolate between states to reconstruct intermediate conformations, validated with case studies on proteins like MurD, which showed hinge-like transitions consistent with experimental data.
7. ProtSCAPE outperformed traditional graph-based methods (GNNs) in predicting pairwise distances and dihedral angles, demonstrating its superior ability to decode the dynamics of protein conformations.
8. This tool holds significant promise for studying complex protein functions, such as allostery, binding, and enzymatic catalysis, by providing a comprehensive view of protein motion across various temporal scales.
@egbertcastro@dbhaskar92@KrishnaswamyLab@Siddharth2814
💻Code: https://t.co/HyCXlNjY0x
📜Paper: https://t.co/6U6U1G9SY0
#ProteinDynamics #DeepLearning #Bioinformatics #MolecularDynamics #Transformer #ProteinConformation #MachineLearning #ComputationalBiology