I’ll be presenting our #GenerativeAI drug design method DrugHIVE at #ACS tomorrow. Come check it out! @JCIM_JCTC
talk: Automated drug design and optimization with structure-based generative deep learning
paper: https://t.co/QsVvHrQOd5
code: https://t.co/PiOrNYczSd
Since the 1960s, the genetic code has been used to predict protein sequences from DNA and mRNA sequences. Our @Nature article demonstrates that these predictions miss thousands of protein sequences present in human tissues.
Across >1,000 human samples, we identified numerous abundant proteins whose amino acid sequences differ from those predicted by the genetic code.
These proteins are not rare translation byproducts. They accumulate to thousands of copies per cell. Some are more abundant than the proteins predicted by the genetic code from the same transcripts.
Their abundance reflects a combination of alternate RNA decoding mechanisms — including codon-anticodon mismatches, tRNA abundance, and RNA modifications — and selective stabilization of the resulting proteins. The last factor – protein stability – emerges as a major determinant of protein abundance across proteins, proteoforms and cell types: https://t.co/IzOfAZKnxT
Alternate RNA decoding is pervasive across functional groups of proteins, healthy and diseased tissues. It affects proteins playing key roles in neurodegeneration, and some alternately decoded proteins show strong enrichment in tumors compared to their surrounding tissues.
This discovery has been a long and exhilarating journey with Shira Tsour and the @slavovLab team. It started in 2019 and proceeded through many challenges and thrilling highs. A journey that has opened new perspectives that we long to explore!
1/
Big news from Boltz - our biggest update yet! 🚀
Today we’re releasing two new state-of-the-art models for protein and small molecule design with extensive wet lab validation and a new API to run all of our models on scalable GPUs wherever you (or your agents) work! 🔥
🍹Weekend project: I (with help from claude - budget $40), was able to get OpenFold3 weights running inside AlphaFold3 codebase (jax). Works for proteins, ligands, rna/dna (1/3)!
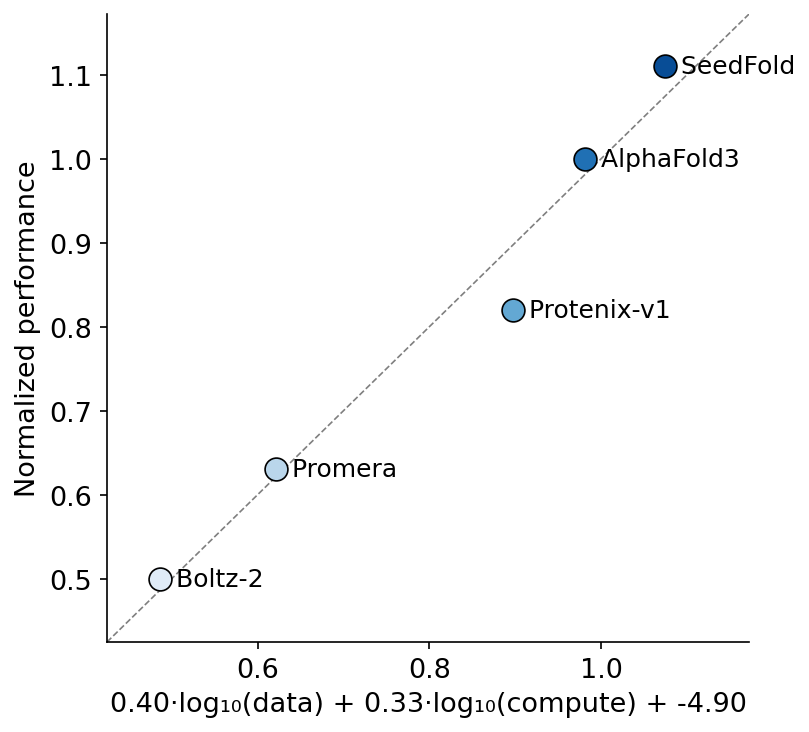
We also found a scaling law that explains (p=0.03) Ab-Ag accuracy across model lineages - from Boltz to AF3 and beyond. 1.8x-ing AF3 performance (to match IsoDDE) would seem to require e.g. 10x more distillation and 16x compute. (7/8)
@mihirbafna14 and I are excited to introduce Promera, a co-folding and design model with
• best-in-class binder filtering
• nanobody design with in-silico success rates matching hallucination
• case studies on hantavirus epitope targeting and GPCR agonism (1/8)
Structural motif search across the protein-universe with Folddisco @NatureBiotech
1 Folddisco is a structural-motif search engine designed for the “protein structure explosion” era: it can query 53 million clustered AlphaFoldDB structures (AFDB50) in seconds, enabling large-scale discovery of short 3D functional patterns (active sites, binding motifs, interfaces) that are often conserved beyond sequence similarity.
2 The key engineering idea is a compact inverted index over position-independent, pairwise geometric “feature encodings” derived from proximal residue pairs (<20 Å). Unlike prior motif indices that store positions, Folddisco stores only structure IDs (with delta compression), shrinking storage while keeping lookups fast.
3 Folddisco extends the classic RCSB-style residue-pair representation by adding two side-chain orientation features: dihedral angles (N1–CA1–CB1–CB2 and N2–CA2–CB2–CB1). These torsion features improve sensitivity and reduce an overly permissive prefilter (ablation reduces F1 and increases runtime).
4 Querying is a 4-stage pipeline: (i) extract/encode query pair features, (ii) prefilter candidates by index lookup (including tolerance-based “extended search”), (iii) residue matching via a compatibility graph and connected components, (iv) superposition and scoring (RMSD plus optional TM-score/GDT/Chamfer/Hausdorff).
5 Candidate ranking is driven by a rarity-aware coverage score: shared feature encodings are weighted by inverse document frequency (IDF), rewarding rare motif-defining geometry and down-weighting ubiquitous patterns (e.g., helix-like encodings). A length penalty helps avoid random matches in large proteins and keeps behavior consistent from tiny motifs to longer fragments.
6 Scale results: Folddisco builds an index for 53M AFDB50 structures in <25 hours with a 1.45 TB index, reported as ~11x faster to build and ~4x smaller than state-of-the-art pair-index approaches; querying is ~20x faster than pyScoMotif (and prefilter-only can be ~100x faster), with AFDB50 prefilter taking ~12 seconds.
7 Accuracy highlights: on human-proteome motif benchmarks, Folddisco notably improves recall for a full 4-residue C2H2 zinc-finger motif where RCSB/pyScoMotif show low sensitivity; for longer discontinuous segment queries, Folddisco outperforms MASTER while being much faster and using a much smaller index.
8 Generalization benchmarks: on SCOPe-derived “scattered conserved residue” queries (5,753 motifs), Folddisco shows substantially higher sensitivity than pyScoMotif (AUC 0.837/0.732/0.504 vs 0.285/0.290/0.300 for full/60%/20% conserved-column sampling). On M-CSA catalytic-site retrieval, Folddisco improves AUC over pyScoMotif (0.432 vs 0.344; a tuned “Sensitive” mode reaches 0.463).
9 Example applications shown: (i) motif-based functional annotation in sequence-divergent/uncharacterized proteins (including metagenomic and non-model-organism hits), (ii) distinguishing GPCR activation states using state-defining motifs (CWxP, NPxxY, DRY), (iii) cross-chain protein–protein interface motif search (immunoglobulin-like interfaces), plus demonstrations of double-motif querying, disulfide/knottin motif detection, and even short linear motif patterns.
10 Limitations discussed: the connected-component matching and 20 Å proximity constraint can miss widely separated multi-site motifs (e.g., distant allosteric pockets), fixed binning can lose borderline matches, and IDF coverage ranking is not always optimal for very short motifs (RMSD ranking can help there). The authors propose motif-specific E-values and variable binning, and aim to extend to nucleic-acid and ligand motifs.
💻Code: https://t.co/C30tYEHTo4; https://t.co/LKzM4LzuM3
📜Paper: https://t.co/BNikAEL3tN
#computationalbiology #bioinformatics #structuralbiology #proteins #alphafold #motifsearch #proteinfunction #enzymes #GPCR #proteininterfaces
super excited to share our latest work! are we really tilting? 🤨
tldr: reward guidance for flows and diffusions is supposed to sample from the reward-tilted distribution. we show it doesn’t 😰 and how to (mostly) fix it ✨
plus lots of fun images!! 🖼️
collaboration with the awesome @nmboffi
website: https://t.co/nvOaAiGYq1
paper: https://t.co/EtkeyiuX7s
code: https://t.co/V3Bi4IVPbf
I’m so excited about the launch of ESMFold2, ESMC, and the new ESM Atlas. This was a massive team effort, and I’m grateful to have worked with such an incredible group @biohub.
A headline result I’m especially excited about: ESMFold2 can design minibinders and antibodies with nanomolar affinity, target selectivity, and functional activity against therapeutically relevant targets.
Today, we’re sharing the full binder design protocol.
Binder design has come of age thanks to generative models—but how can we access the wider array of dynamic, multistate protein functions, so elegantly employed by nature?
@mihirbafna14 and I are excited to share SwitchCraft, a framework for designing such functions. (1/7)
#MeteorSighting: Eyewitnesses in New England and @NOAA’s GOES-19 satellite reported a bright fireball on Saturday, May 30, at 2:06 p.m EDT accompanied by a loud noise. The meteor appears to have fragmented at an altitude of 40 miles over northeast MA and southeast NH. The energy released at breakup is estimated to be equivalent to about 300 tons of TNT, which accounts for the loud noise.
Eyewitness accounts supplied by the American Meteor Society.
Reports of an explosion hears around Boston I believe are going to be a rather significant bolide/meteor entering the atmosphere. Very large "flash" detected by GOES-19 GLM that does not correlate with active thunderstorms. #MAwx
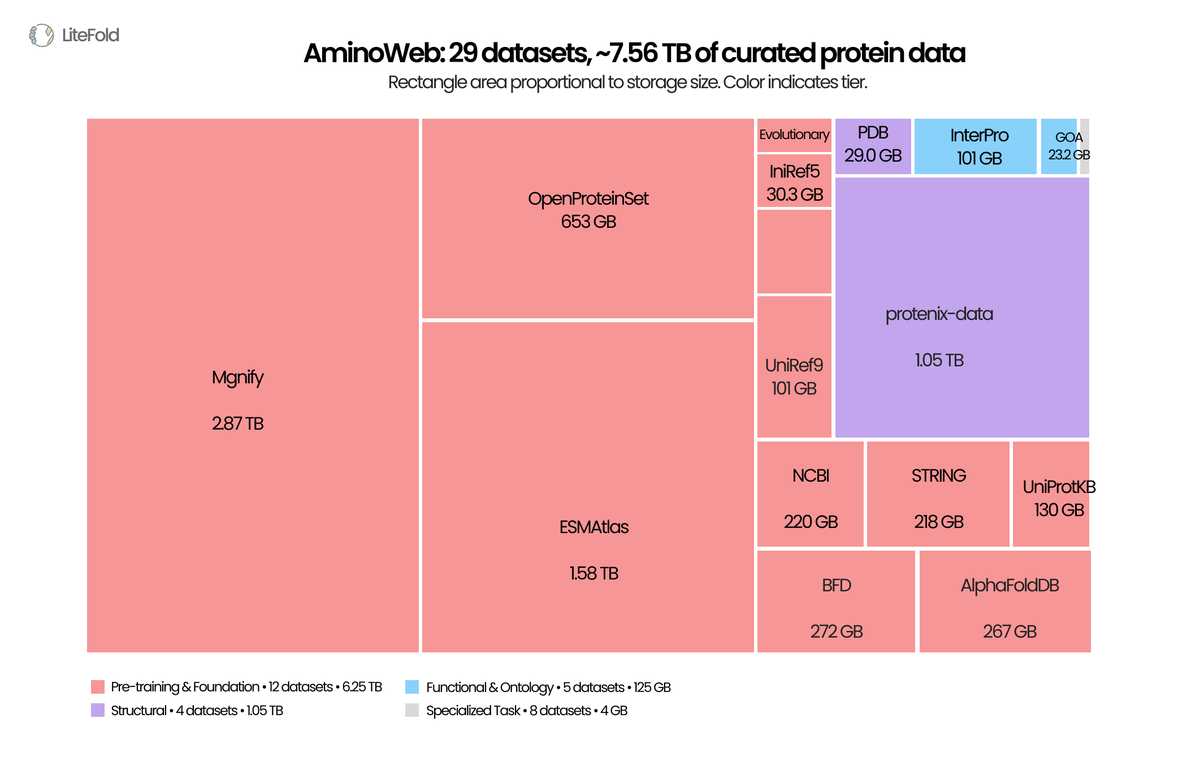
LLMs got FineWeb, The Pile, RedPajama, Dolma. Protein ML got per-paper supplementary tables and FTP mirrors scattered across a dozen institutions.
Today we're releasing AminoWeb on @huggingface : 29 cleaned, ML-ready protein datasets, ~7.5 TB total. Sequence, structure, function, MSA, variant-effect, stability, binding. UniProt, PDB, AlphaFoldDB, ESMAtlas, ProteinGym, MegaScale, Protenix, and more.
Typed Parquet. Homology-aware splits. Preserved score conventions. Full provenance per record.
Protein ML scaled architectures for years while the data layer stayed fragmented. We've also shared the full curation pipeline, case studies, and observations in the companion blog post.
Access the data: https://t.co/elQ7pzpNkG
Read the release blogpost: https://t.co/28yFU2m9Jc
Today we're announcing ESMFold2, an open scientific engine to power prediction, design, and discovery across protein biology.
The new model delivers state of the art performance on protein interactions, especially antibodies, a critical modality for therapeutics.
We have designed and validated miniprotein binders and single chain antibodies across five therapeutic targets that are important in cancer and immunology. We are seeing very high success rates, and affinities at levels consistent with therapeutic activity.
We’re also releasing an atlas of 6.8 billion proteins, and 1.1 billion predicted structures.
ESMFold2 is built on a state of the art language model that has been trained on billions of protein sequences.
A world model of protein biology emerges through language modeling.
We’ve used the techniques of mechanistic interpretability developed to understand large language models to understand the concepts ESM uses to represent proteins.
The model’s representation space has a compositional organization of features across scales, levels of complexity, and abstraction, that reflects and mirrors the understanding of protein biology developed through a century of empirical science.
This understanding emerges without prior knowledge, just from language modeling of protein sequences.
Language models are becoming a powerful substrate to understand and program biology.
The design of protein interactions is one of the most fundamental problems in biophysics, and has critical implications for the discovery of new medicines. A simple gradient based search with the model was able to discover high-affinity protein binders.
I'm excited by the potential this has to accelerate basic science and the understanding of proteins. And especially for the new avenues it opens up for therapeutic design and medicine.
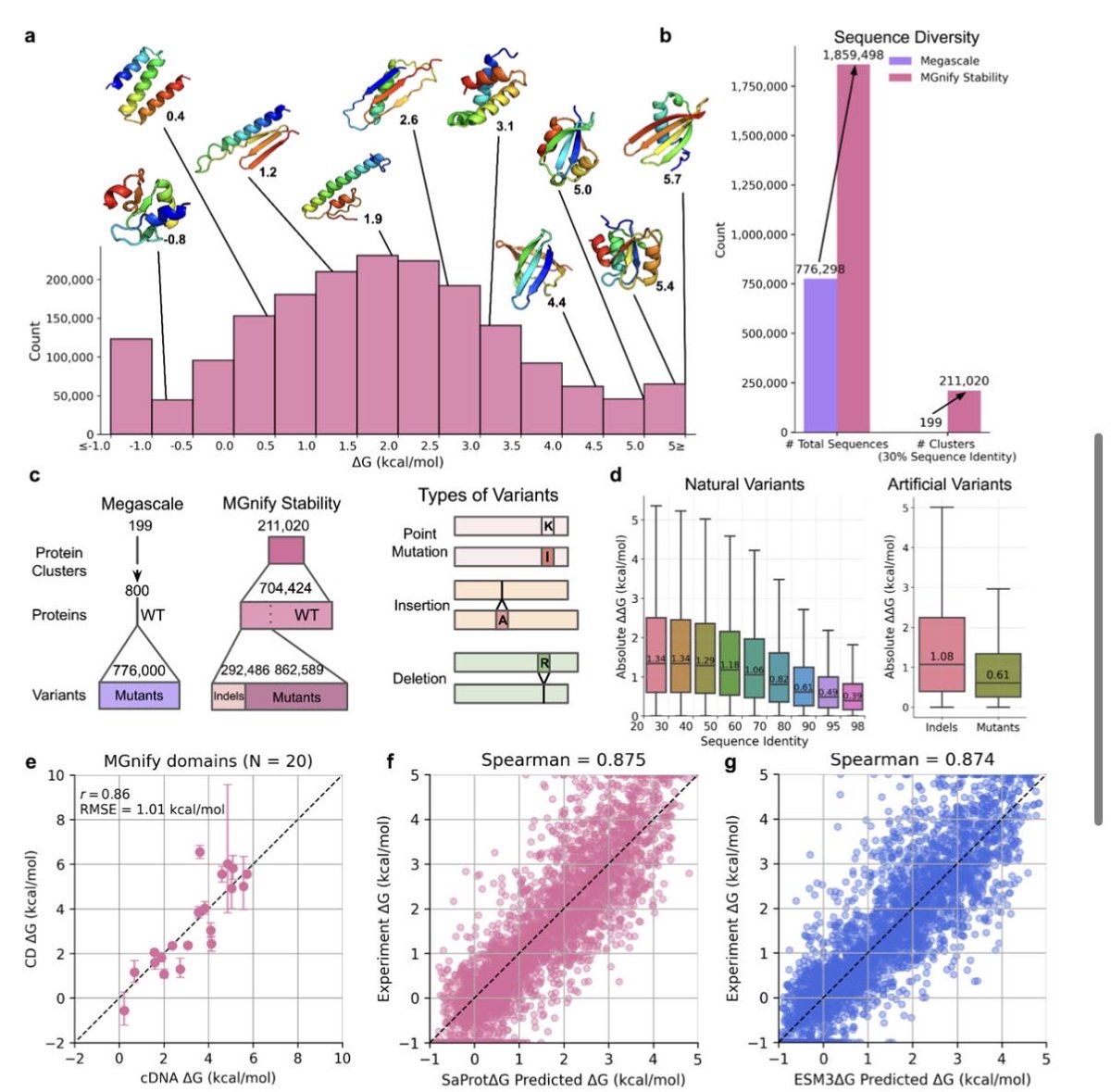
❗️Accurate protein stability prediction for small domains using mega-scale experiments ❗️
https://t.co/VCNkMeTRb7
+1.8 million protein stabilities measured. High quality data in, high quality model out!
Congrats @ChoYehlin@KotaroTsuboyama@grocklin and whole team!
📢Excited to share our new paper:
Continuous Diffusion Scales Competitively with Discrete Diffusion for Language
We introduce RePlaid 🌊, a continuous diffusion language model (DLM) with
🏅Discrete likelihood bound
🏅Scaling laws competitive with SOTA discrete DLMs
How? Dive in👇[🧵1/12]
Paper: https://t.co/Q00fmvtHmy
Work done with my amazing collaborators: @WeiGuo01@ShuibaiZ69721@ssahoo_@YongxinChen1@ArashVahdat@MardaniMorteza@jwthickstun
Thrilled to share our new paper where we introduce a multiplexed hydrogen–deuterium exchange MS (mHDX‑MS) method that can measure hundreds of protein domains’ conformational energy landscapes—all in a single experiment!
https://t.co/E6QfRWiDVf
I’ve always believed the No.1 application of AI should be to improve human health.
That work started with AlphaFold, and now at @IsomorphicLabs with the mission to reimagine drug discovery and one day solve all disease!
We are turbocharging that goal with $2.1B in new funding.
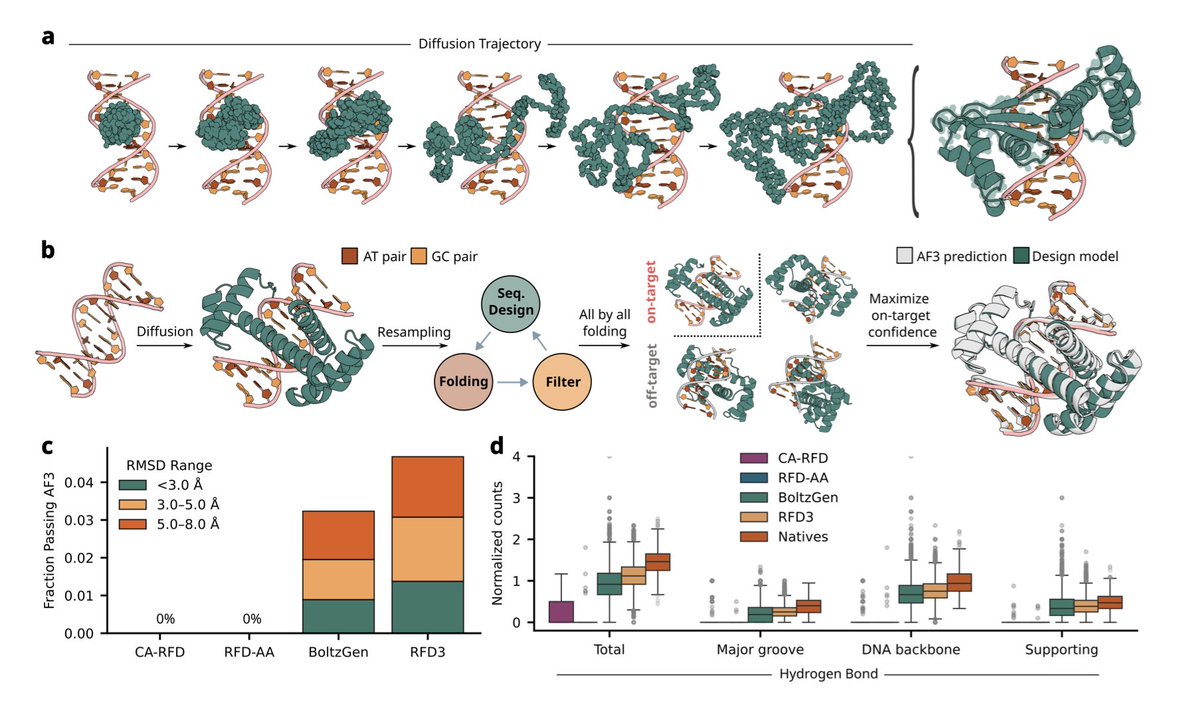
Generative Design of Sequence Specific DNA Binding Proteins
🚀 New preprint from David Baker!🚀
1. This study achieves a ~100-fold improvement in success rates for de novo DNA binder design compared to previous approaches, identifying specific binders for 7 out of 15 diverse DNA targets by testing only 96 designs per target.
2. The method combines RFdiffusion3 for structure generation with explicit AlphaFold3-based screening against off-target interactions, addressing the fundamental challenge that B-form DNA has similar global structures across sequences with only subtle chemical differences between bases.
3. The design pipeline consists of two phases: a "binder block" focused on generating proteins with dense atomic interactions with target DNA, and a "specificity block" that filters designs by comparing AlphaFold3 confidence between on-target and off-target predictions using ΔminPAE metrics.
4. Key innovations include maximizing hydrogen bonding networks with DNA bases for direct readout and phosphate backbone contacts for indirect readout, alongside preorganized side-chain conformations stabilized by intraprotein buttressing interactions to reduce off-target binding.
5. Experimental validation using yeast surface display revealed that in silico specificity filtering boosted specific design frequency by 6-fold (from 0.5% to 3%), with most binding designs from the specificity block also being sequence-specific.
6. Purified designs showed nanomolar binding affinities (KD 3-30 nM), and single-base resolution competition assays demonstrated robust sequence discrimination across diverse targets, with designs often preferring intended targets over 35 out of 40 single base variants.
7. The designed proteins are structurally novel with no close global matches to known DNA-binding proteins in the PDB, yet achieve binding affinities comparable to native transcription factors, representing genuinely de novo solutions rather than redesign of existing scaffolds.
8. Structural diversity among successful designs includes both modular helical bundle architectures reminiscent of homeodomains and integrated monolithic folds distinct from natural transcription factors, highlighting the broad solution space accessible to generative diffusion models.
9. The study identifies important trade-offs between folding and selectivity, with more stringent off-target filtering reducing expression rates, suggesting future improvements could come from direct specificity optimization during inference rather than post-hoc filtering.
10. Applications span programmable gene regulation, targeted genome manipulation, and synthetic regulatory systems, with targets including therapeutically relevant sites for PRNP, PCSK9, DUX4, and the TATA box for which no natural major groove binder exists.
💻Code: https://t.co/uOa9TDJlw2
📜Paper: https://t.co/cZBpaCRheg
#DeNovoProteinDesign #DNABindingProteins #RFdiffusion3 #AlphaFold3 #ComputationalBiology #ProteinDesign #GenomeEngineering #SyntheticBiology #TranscriptionFactors #DeepLearning
Generative design of sequence specific DNA binding proteins.
Most fun paper I have ever written, with @enishasehgal and @YPolitansk15183 and the team, on a project which is a testament to the power of RFdiffusion3.
https://t.co/iYmdXRDBhQ





















![zhihanyang_'s tweet photo. 📢Excited to share our new paper:
Continuous Diffusion Scales Competitively with Discrete Diffusion for Language
We introduce RePlaid 🌊, a continuous diffusion language model (DLM) with
🏅Discrete likelihood bound
🏅Scaling laws competitive with SOTA discrete DLMs
How? Dive in👇[🧵1/12]
Paper: https://t.co/Q00fmvtHmy
Work done with my amazing collaborators: @WeiGuo01 @ShuibaiZ69721 @ssahoo_ @YongxinChen1 @ArashVahdat @MardaniMorteza @jwthickstun](https://pbs.twimg.com/media/HIpwk2wXkAA7ebW.jpg)