Much ado about nothing: Modeling amino acid replacement with predicted protein structures
1 The paper introduces AFSM (AlphaFold Substitution Matrix), built BLOSUM-style but from predicted 3D structures: >20,000 proteins across 297 InterPro families, yielding ~660,000 structural alignments and ~68.4M residue pairs to estimate amino-acid replacement scores.
2 AFSM construction: within each family, protein pairs were filtered to avoid redundancy (>62% identity removed), structurally aligned, residue correspondences extracted from spatial proximity (ungapped), family contributions weighted to reduce large-family bias, then converted to half-bit log-odds scores and rounded like BLOSUM62.
3 Despite using structural data at much larger scale than classic matrices, AFSM ends up very close to BLOSUM62: 48% of entries identical; most other entries differ by only ±1 (largest differences around His/Cys). Overall correlation with BLOSUM62 is 96%, and AFSM’s “evolutionary distance” (match/mismatch ratio summary) is closest to BLOSUM62.
4 Benchmarking spans five tasks: two MSA reconstruction benchmarks (BAliBASE R10, 209 MSAs; plus a large Pfam Seed subset, 18,061 MSAs) and three remote homology search settings (CATH, SCOPe40, and the hardest CATH S20 with 20% identity filtering).
5 Main empirical result: AFSM and BLOSUM62 perform nearly identically across all five benchmarks. Example metrics (percent): BAliBASE Q-score 71 vs 70; Pfam Seed Q-score 82 vs 81; homology ROC AUCs essentially match (CATH 87/87, SCOPe40 79/80, CATH S20 64/64), with similar optimal gap penalties after per-matrix tuning.
6 Extending beyond AFSM vs BLOSUM62, the study compares 16 additional matrices (PAM, VTML, Gonnet, PFASUM, RBLOSUM, CORBLOSUM, SDM/HSDM, ProtSub). Across MSA and homology tasks, most matrices fall within a narrow performance band; neither publication year, data size, nor “structure-derived vs sequence-derived” reliably predicts better results.
7 A notable control: very simple “baseline unit matrices” (match score 5; mismatch score 0 or -1) can match established matrices on MSA reconstruction, implying that much of the benefit comes from a generic conservation signal plus gap modeling rather than fine-grained substitution preferences. However, for homology search, the “no-matrix” style baseline (5/0) degrades more noticeably.
8 The paper links matrix usefulness to MSA size: when MSAs have few sequences, BLOSUM62/AFSM can outperform “no matrix”; when MSAs have many sequences (around >20), they can become slightly worse than the baseline. Interpretation: substitution matrices mattered most when sequence data were sparse; dense MSAs provide enough constraints that detailed substitution scoring adds little.
9 In sparse remote homology, embeddings outperform substitution matrices: ProtTrans T5 embeddings with nearest-neighbor search achieve ROC AUC 99/94/90 on CATH/SCOPe40/CATH S20, versus ~99/89/78 for AFSM or BLOSUM62. The gap is largest in the sparsest setting (CATH S20), supporting embeddings as a better extrapolation tool across large evolutionary distances.
📜Paper: https://t.co/mr5rQ3aJUk
#Bioinformatics #ComputationalBiology #ProteinEvolution #SequenceAlignment #MSA #HomologySearch #AlphaFold #SubstitutionMatrix #ProteinEmbeddings #ProteinLanguageModels
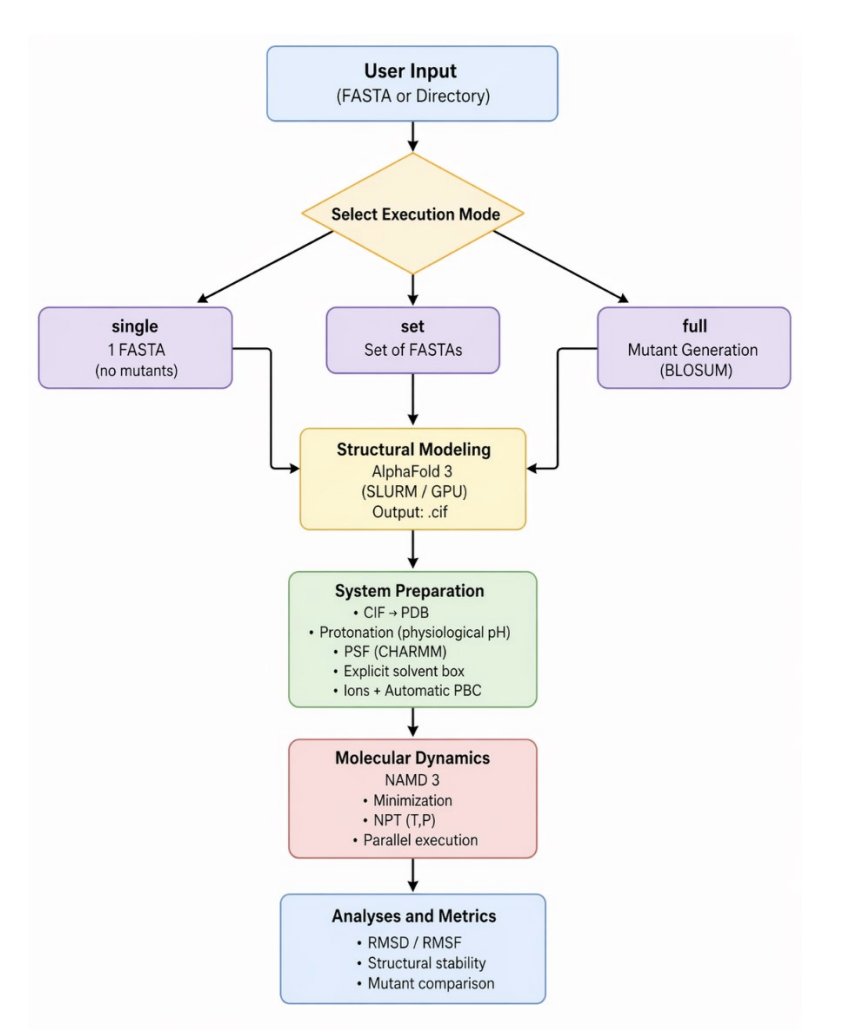
AlphaUnfold: Probing Potential Unfolding and Structural Fragility in AlphaFold3 Models via Short-Time High-Pressure MD
1 AlphaUnfold is an automated “stress-test” pipeline that takes AlphaFold3 (AF3) models and subjects them to short (5 ns) high-pressure MD (1000 atm) in NAMD3, aiming to reveal structural fragility and early unfolding signals that static confidence scores can miss.
2 The key result is a strong inverse relationship between AF3 average pLDDT and MD structural drift: after 5 ns high-pressure MD, lower-confidence models show markedly higher RMSD. The reported regression is RMSD = 35 − 0.34·pLDDT (R² = 0.78, p < 0.001), implying ~3.4 Å less RMSD per +10 pLDDT points.
3 A central validation claim is that 5 ns at 1000 atm captures similar “where it moves” patterns as much longer conventional MD: the study compares high-pressure 5 ns runs to standard-pressure trajectories (notably 200 ns at 1 atm) and finds consistent ranking/patterns of structural drift, with high-pressure runs converging quickly to metastable states (lower RMSD standard error in the final segment).
4 Beyond global RMSD, AlphaUnfold emphasizes local vulnerability mapping: per-residue RMSF profiles align with local pLDDT, where low pLDDT regions (e.g., <50) tend to show high fluctuations (e.g., >5 Å), while high pLDDT regions (>90) remain comparatively rigid (often <2 Å RMSF). This is presented as a practical way to pinpoint metastable segments for redesign.
5 The workflow is designed for throughput and reproducibility: AF3 prediction and pLDDT extraction, automated MD system preparation (solvation/ions/minimization), 5 ns high-pressure production in triplicate, then automated reporting of RMSD/RMSF plus compactness/exposure metrics (RoG, SASA), with optional shape/contact analyses (ellipsoid index, COCaDA contacts).
6 The paper frames the pipeline as particularly useful for protein engineering in single-cell protein (SCP) contexts: when enriching essential amino acids (e.g., via BLOSUM-guided substitutions), AlphaUnfold can serve as a rapid computational filter to reject designs that look plausible in AF3 but are dynamically fragile under stress.
7 The authors highlight important outliers where pLDDT may not predict MD behavior, illustrating how missing chemistry in simulation setup can dominate outcomes: a cited case (Q96M98-2) has high average pLDDT (~85) but unusually high RMSD (11.7 Å), potentially due to omitted stabilizers such as explicitly defined disulfide bonds, or missing cofactors/ligands/ions.
8 The broader message is methodological: pLDDT is informative but static; coupling AF3 with a brief, physics-based perturbation (high-pressure MD) can provide an “experimental-like” robustness check and actionable localization of weak spots, intended to improve confidence in downstream use of AI-generated structures.
💻Code: https://t.co/b1MzVriVCg
📜Paper: https://t.co/bWPQ08hSUX
#AlphaFold3 #MolecularDynamics #ProteinStructure #ComputationalBiology #ProteinEngineering #NAMD #StructuralBioinformatics #HighPressureMD #SCP
1/ Why do some genetic diseases remain unsolved even after whole-genome sequencing? The 5′UTR could hold the answer. In @AJHGNews, we present 5ULTRA, a new computational method to decode how these variants affect protein translation.
https://t.co/sE3sl9mrZZ
Steering semi-flexible molecular diffusion model for structure-based drug design with reinforcement learning @ScienceAdvances
1. The paper introduces SeFMol, a reinforcement learning (RL)–steered 3D diffusion model that treats diffusion denoising as a Markov decision process, enabling semi-flexible, stepwise conformational adjustment of ligands inside protein pockets rather than assuming rigid ligand conformations.
2. Core idea: a pretrained “rigid” conditional diffusion model is further optimized with a semi-flexible RL stage (SFRL). A frozen reference denoiser anchors the policy denoiser via a KL constraint, helping prevent reward overfitting and catastrophic drift while improving binding-oriented geometry.
3. Property control is built into generation as explicit conditioning signals (computed with RDKit): QED, SA, LogP, TPSA, HBA, HBD, Fsp3, and ROTB. These conditions act as dense guidance throughout denoising to counter sparse terminal rewards from docking-based objectives.
4. Training pipeline: (i) pretrain on 1,000,000 target-free Molecule3D molecules with property conditioning to learn general structure–property priors; (ii) fine-tune on 100,000 CrossDocked2020 protein-ligand pairs to become pocket-aware while retaining property bias; (iii) apply SFRL to steer denoising trajectories toward better pocket complementarity.
5. Efficiency contribution: a variable fast sampling strategy reduces diffusion steps from 1000 (training) to 50 (sampling/optimization), yielding ~20x fewer steps while maintaining quality; reported sampling is ~0.81 s per molecule with 98.3% completion, substantially faster than several diffusion baselines.
6. Docking-centric results on 100 test pockets (100 molecules per pocket): SeFMol reports average Vina score −7.23 kcal/mol and improved affinity-related metrics (Vina min and Vina dock). It also reports an SR (success rate under nine joint affinity+property constraints) of 11.53%.
7. Geometric/interaction reliability: SeFMol’s generated poses show strong agreement between direct Vina scoring and redocking (reported correlation 0.95), plus competitive RMSD-to-redocked distributions and fewer docking clashes, consistent with the goal of generating chemically plausible 3D conformations directly.
8. Interaction-pattern preservation with optimization: using PLIP interaction typing, SeFMol maintains interaction-type distributions close to reference ligands (reported JSD 0.1401, comparable to the best baseline), suggesting affinity gains do not come from unrealistic interaction artifacts.
9. Generalization case studies: on real-world targets CDK2 (1H00) and ROCK1 (6E9W), SeFMol-generated ligands reproduce canonical interactions seen in known actives while also proposing alternative chemotypes and plausible new interactions; property steering (e.g., setting TPSA or Fsp3 targets) shifts generated distributions around specified values.
📜Paper: https://t.co/DeVNTc4KoQ
#ComputationalBiology #StructureBasedDrugDesign #DiffusionModels #ReinforcementLearning #MolecularGeneration #GenerativeAI #DrugDiscovery #GeometricDeepLearning #SE3Equivariance
Does AlphaFold’s latent space encode only the native state or something like a distribution over conformations? We begin to answer this question with ConforNets, a mechanism for producing diverse states, or very specific ones, via inference-time adaption of OF3p’s latent space👇
Join us for a joint webinar with IDT’s Principal Scientist Kim Lennox, MSc, and n-Lorem’s Assistant Director of ASO Strategy Research, Anthony Vu, PhD who explore ASOs in personalized medicine.
Register here: https://t.co/sn5VTsIw26
A drug that targets muscarinic acetylcholine receptors recently became the first in a new class of treatments for schizophrenia in decades - read more about targeting this GPCR family in this review
https://t.co/rY51NSRv6L
https://t.co/wFt3l91l5R
Approximately 13% of Hispanics who are 65 or older have Alzheimer's or another dementia.
This #MinorityHealthMonth, learn how Alzheimer’s affects the Hispanic American community at https://t.co/ZGP3SlwZ4v
Boltz-2 just got a major speed upgrade. 🚀
We’re releasing Lightning-Boltz, a local, GPU-accelerated framework free from public MSA server bottlenecks.⚡
On a single L40S, total runtime drops to 28s per input vs 89s with the rate-limited server and 298s with MMseqs-CPU.
1/5 🧵
Download the 2025 Annual Report for an overview of recent activities and how RCSB PDB supports the broad PDB user community with a wide range of resources understanding 3D biostructures
https://t.co/acVOgYpE1M
Tune in on Tuesday, April 28 at 3 PM EST to hear Juan Pablo Tosar, PhD
“Beyond vesicles: uptake and sensing of inflammatory extracellular RNAs”
To join virtually, register through this link: https://t.co/VnKhJxiVpE
#RNA#RNAMedicine@BIDMC_CancerCtr
A limited number of poster presentation slots remain for RNA 2026 in Montréal, Canada from May 26th – 31st.
We encourage you to register and submit your poster abstract by April 24th to secure one of the remaining spots!
https://t.co/GQA8HS8ovU
LinkLlama: Enabling Large Language Model for Chemically Reasonable Linker Design
1. LinkLlama reframes fragment linking as instruction-following SMILES generation with explicit geometric constraints (fragment anchor distance in Å and angle in degrees) plus optional property bands—aiming to avoid the strained, non-drug-like linkers often produced by purely 3D generative methods.
2. The key design choice is “alignment-by-design”: instead of reinforcement learning loops, the model is supervised fine-tuned to internalize medicinal-chemistry constraints directly, then steered at inference time via natural-language prompts.
3. Training data comes from a large, curated ChEMBL36 pipeline: ~2.67M cleaned drug-like molecules are fragmented (two cuts via RDKit MMPA) into 8.3M fragment–linker–fragment triplets (256k unique linkers), with strict rules to keep linkers realistic (e.g., cuts on acyclic single bonds incident to neutral sp3 carbons; minimum sizes; linker not dominating fragments).
4. “Chemical reasonability” is operationalized with a stringent 5-part filter set: linker-level checks (bridgehead/overly complex ring structures; uncommon ring systems <100 occurrences in ChEMBL) and molecule-level checks (iMiner undesirable SMARTS, PAINS, Brenk). A molecule is “reasonable” only if it passes all five.
5. The model is Meta Llama-3.2-1B-Instruct fine-tuned with LoRA on 1.5M rebalanced examples (Cap50 per-linker frequency cap to reduce memorization of common motifs like amides). Training is lightweight (reported ~6.5 hours on 4×A100), emphasizing practicality.
6. A notable modeling twist: the output is a JSON containing both the proposed linker SMILES and a “reasoning” trace that explicitly reports pass/fail status for the same chemical filters—teaching the model to co-generate structures and their medicinal-chemistry compliance.
7. On standard ZINC benchmarks (random 1k + hard 1k), LinkLlama achieves near-perfect validity (~99.9%) and competitive uniqueness, while substantially increasing chemically reasonable designs: reasonability rises to ~73% (random) and ~87% (hard), vs ~44% for DeLinker and ~25–31% for DiffLinker.
8. 3D evaluation shows a key tradeoff improvement: fragment pose agreement (RMSD) is broadly comparable across methods, but LinkLlama’s MMFF strain proxy (ΔE) avoids the heavy tail seen in the 3D diffusion baseline, suggesting fewer distorted/high-strain linker geometries even though LinkLlama generates in SMILES space.
9. Prompt-conditioned control is a central capability: on ZINC hard 1k, LinkLlama can satisfy combined constraints (e.g., ring-containing + Lipinski Ro5 + minimum linker size/rotatable bonds + “reasonable”) with success rates in the ~40% range, while baselines and unconditional sampling collapse to near-zero under the strict joint constraints.
10. Prospective-style case studies extend beyond benchmarks: scaffold hopping on mineralocorticoid receptor (PDB 6L88) yields alternative heterocyclic cores with stable 200 ns MD behavior; PROTAC linker redesign on BRD4–VHL (PDB 6SIS) generates linear linker alternatives to a macrocyclic architecture, with MD suggesting comparable or improved ternary complex stability.
💻Code: https://t.co/mgQMHDjfzR
📜Paper: https://t.co/pynfL0eEe0
#ComputationalChemistry #DrugDiscovery #FBDD #GenerativeAI #LLM #Cheminformatics #PROTAC #MolecularDesign #MachineLearning
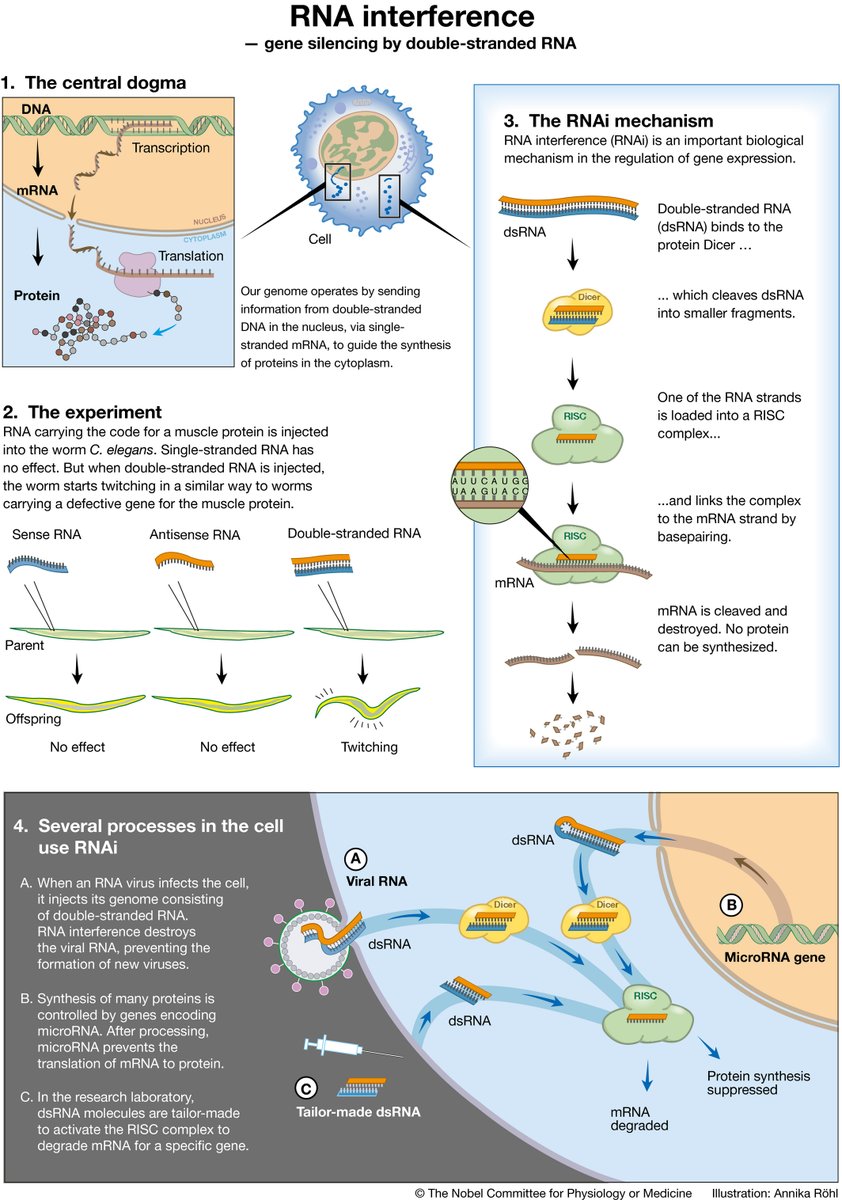
A strand of RNA has been discovered to perform two key reactions required for self-replication. The molecule offers a clue towards understanding the origin of life, namely how non-living chemistry on prebiotic Earth started to self-replicate and evolve.
https://t.co/3bsnGMSZry