🚨New paper! Generative models are often “miscalibrated”. We calibrate diffusion models, LLMs, and more to meet desired distributional properties.
E.g. we finetune protein models to better match the diversity of natural proteins.
https://t.co/2c06vD0x2D
https://t.co/9Tbhf6ml8K
A Chemoenzymatic Cascade for the Formal Enantioselective Hydroxylation and Amination of Benzylic C−H Bonds; Y. Zhang, C. Huang, W. Kong, L. Zhou, J. Gao, F. Hollmann, Y. Liu,
& Yanjun Jiang #singleatomcatalysts#peroxygenase#enantioselective https://t.co/R2Dlimws9K
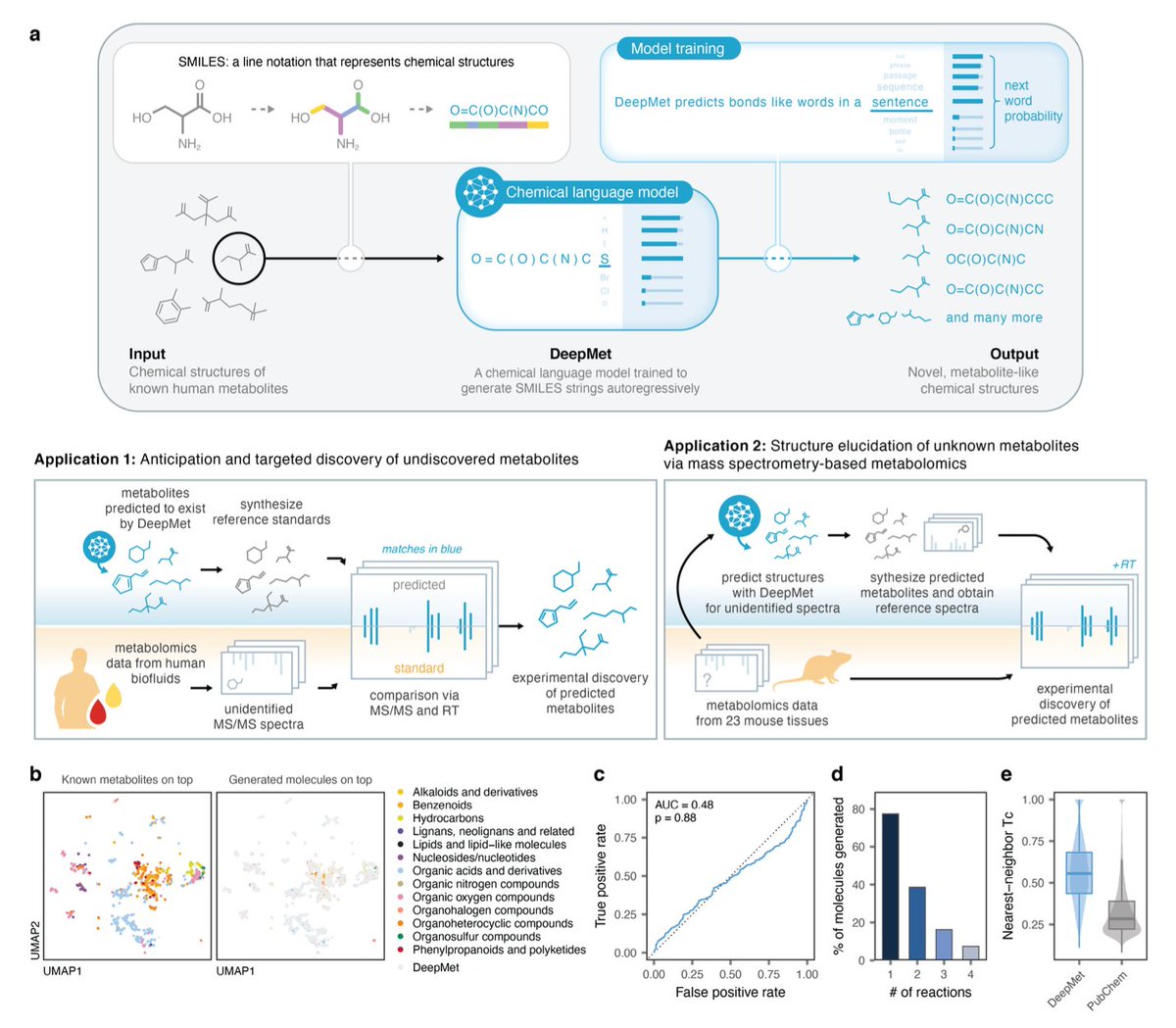
Language model-guided anticipation and discovery of unknown metabolites
• The study introduces DeepMet, a chemical language model designed to anticipate and discover previously unidentified metabolites by learning biosynthetic logic from known metabolite structures.
• DeepMet predicts the structures of novel metabolites and integrates with mass spectrometry (MS/MS) data to systematically discover metabolites within complex biological samples.
• The model demonstrated success in predicting 81% of newly discovered human metabolites from the Human Metabolome Database (HMDB) version 5, highlighting its robust predictive power.
• Through experimental validation, DeepMet led to the discovery of 47 novel mammalian metabolites, showcasing its ability to uncover structurally diverse metabolites spanning various chemical classes.
• Integration with MS/MS workflows significantly improved the identification of metabolites, with DeepMet aiding in the structural elucidation of unidentified peaks in metabolomics data.
• A repository-scale application of DeepMet annotated over 29.1 million MS/MS spectra, substantially increasing the number of matched experimental spectra compared to traditional methods.
• The study highlights DeepMet’s ability to bridge gaps in metabolic databases, enabling high-throughput identification of the “chemical dark matter” in the metabolome.
@skinniderlab@WishartLab@EvolvedChem@BoWang87@AdamoYoung@NeinastM@AsaelRoichman
📜Paper: https://t.co/qZcNFI4fGF
#Metabolomics #ChemicalLanguageModel #Bioinformatics #MachineLearning #MetabolicDiscovery
Amazing collaboration with Stoltz group @Caltech and Davies group @EmoryUniversity! Our part was to achieve tetra-CH hydroxylation via weak coordination at very late stage of complex total synthesis:
https://t.co/vFziz8kmin
🚨 So, why do we need weight decay in modern deep learning? 🚨
The camera-ready version of our NeurIPS 2024 paper is now on arXiv (a major update compared to the first version).
Weight decay is traditionally viewed as a regularization method, but its effect in the overtraining regime is quite subtle and its interaction with the implicit regularization effect of SGD plays a crucial role.
In the undertraining regime (e.g., in LLM pretraining), however, the effect of weight decay is totally different: it sets an implicit learning rate schedule for AdamW and enables stable training with bfloat16 precision. This explains why weight decay is still widely used for LLM training with standard optimizers, such as AdamW.
This is joint work with @dngfra, @adityavardhanv, @tml_lab.
PDBe tools for an in-depth analysis of small molecules in the Protein Data Bank
1. PDBe has developed new tools to facilitate the analysis of small molecules in the PDB, enhancing data accessibility and insight into ligand-macromolecule interactions. The tools—PDBe CCDUtils, PDBe Arpeggio, and PDBe RelLig—enable researchers to explore ligand data with greater depth and accuracy.
2. PDBe CCDUtils provides enriched ligand data by parsing and processing molecular structures, supporting researchers in standardizing ligand identification across PDB entries. This tool also generates high-quality 2D and 3D visualizations of ligands, enabling a comprehensive view of small molecules.
3. PDBe Arpeggio analyzes detailed interactions between ligands and macromolecules, identifying specific contact points such as hydrogen bonds and hydrophobic interactions, which are crucial for understanding ligand binding.
4. PDBe RelLig classifies ligands based on their functional roles (e.g., cofactor-like, reactant-like, or drug-like), helping researchers distinguish biologically relevant molecules from experimental artefacts, thereby supporting accurate interpretation of biological functions.
5. These tools, along with PDBe-KB’s ligand pages, provide an integrated view of ligands in their biological context, making it easier to visualize interactions, assess binding sites, and explore molecular properties in drug discovery and structural biology research.
@preeti_cy@PDBeurope
💻Code:
• PDBe CCDUtils: https://t.co/IT57YpwmyL
• PDBe Arpeggio: https://t.co/MjAWvGs4DQ
• PDBe RelLig: https://t.co/6ATdBrx9wu
📜Paper: https://t.co/hsIWFJyBUZ
#Bioinformatics #PDB #DrugDiscovery #StructuralBiology #LigandAnalysis
Evidence from omics that aging is not a gradual, linear process. The new study, highlighted here, only assessed people up to age 75 among 108 participants with short term follow-up (1.7 yrs). A previous large study (N>4,200, up to age 95) found a 3rd peak at age 80.
https://t.co/ZSB1AMIgtW
https://t.co/1yCEP11Wav
Delighted to share our latest work on a Chemoenzymatic Diversity-Oriented Synthesis strategy for generating complex, natural-product-like compounds with enzymes. Big 👏to the lead authors Andrew Bortz and John Bennett for this beautiful work!🎉https://t.co/45xnT3n2tJ
Three of my favorite papers published this week:
1. A mechanism for bacteria to create *new* repetitive toxic genes to kill themselves in response to infection (!!)
Design of intrinsically disordered protein variants with diverse structural properties | @ScienceAdvances
- Design new IDP sequences using Simulated Annealing with a target radius of gyration (compaction)
- Initialize with a natural IDP sequence, and swap two random residues at each step (to keep the same composition)
- Evaluate compaction using either MD simulation with CALVADOS or by reweighting prior generated conformations
Link: https://t.co/JrAx04I2Yd
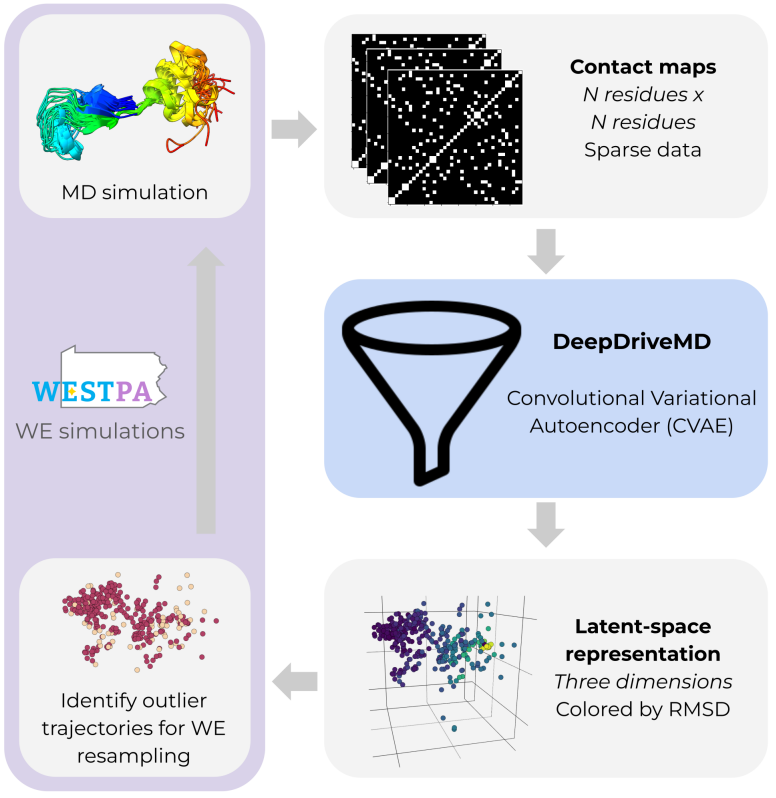
Unsupervised learning of progress coordinates during weighted ensemble simulations: Application to millisecond protein folding
- Improve rare events in protein folding (e.g., state transitions) through weighted ensemble simulation and an unsupervised deep learning model.
- Use a convolutional VAE to compress contact maps into latent space, and applies a Local Outlier Factor to identify outlier conformations, which are then replicated in the simulation.
- Training the CVAE on-the-fly works better than using a pretrained model.
Preprint: https://t.co/gcKKFubk2P
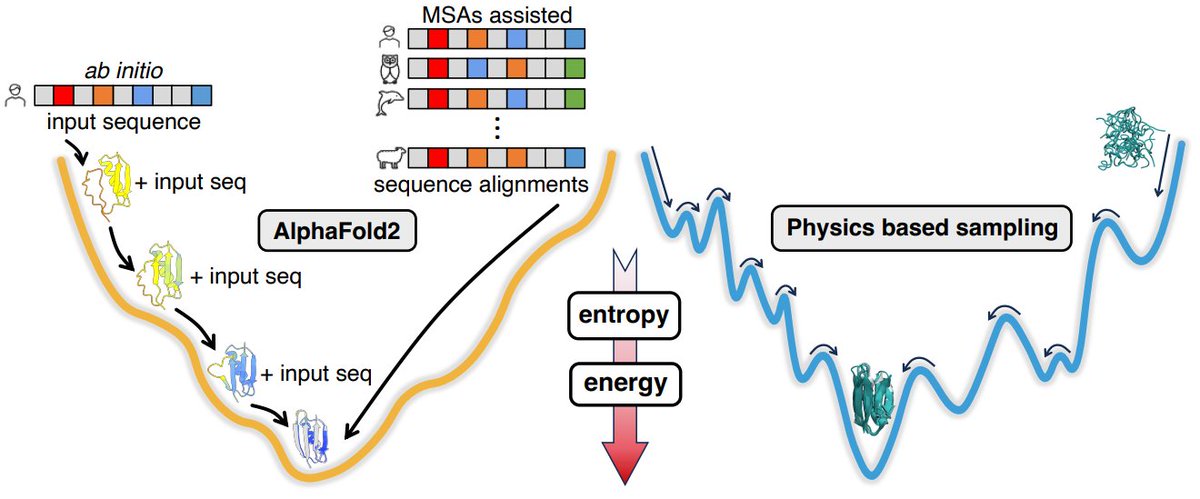
AlphaFold2 knows some protein folding principles
-Use AF2 without MSAs/templates, mimicking an ab initio approach. The iterations show AF2's energy landscape and "local first, global later" folding mechanism.
- Folded intermediates of six small proteins (protein G, protein L and their mutants, ubiquitin, and the SH3 domain) resemble results in other studies and MD simulations.
- Scale iterative folding study with 7k high-resolution proteins clustered by MMSeq2, with lengths ranging from 30 to 250
Preprint: https://t.co/dR1hmuPNMf
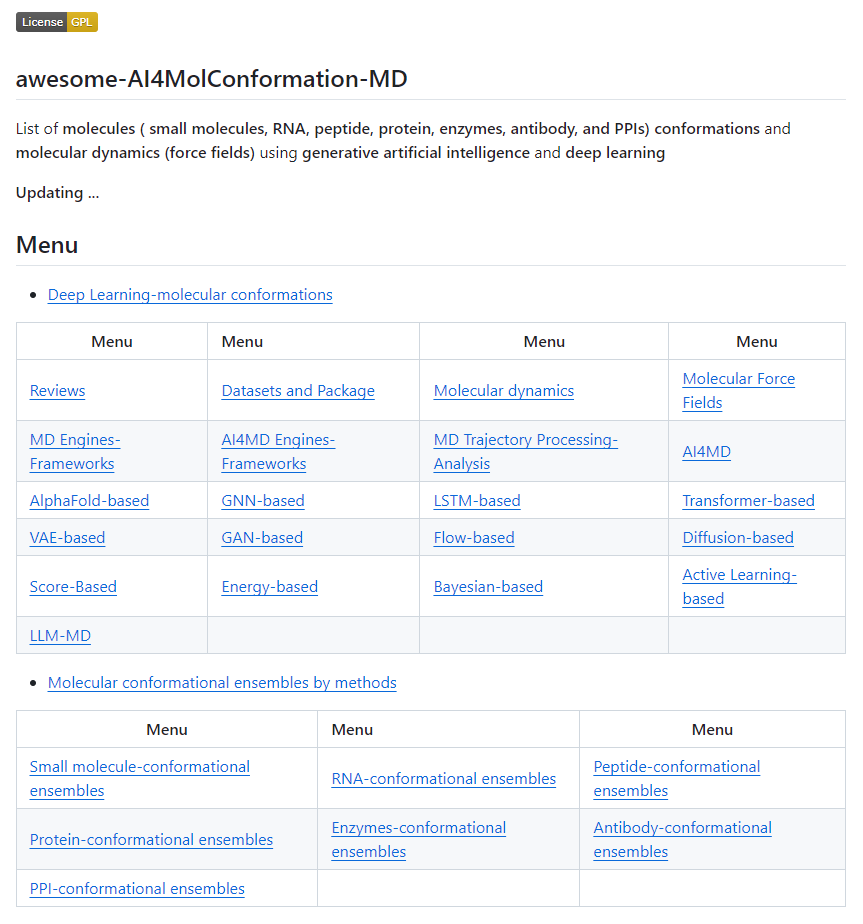
Updating !!!
Collection(GitHub): Deep Learning and Generative AI for molecules ( small molecules, RNA, peptide, protein, enzymes, antibody, and PPIs) conformations and molecular dynamics (force fields)
https://t.co/uc7fGSZPUX
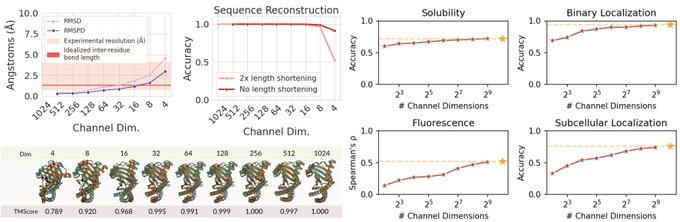
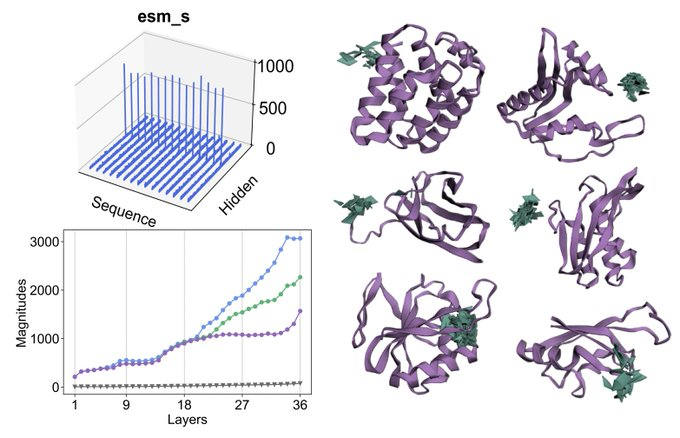
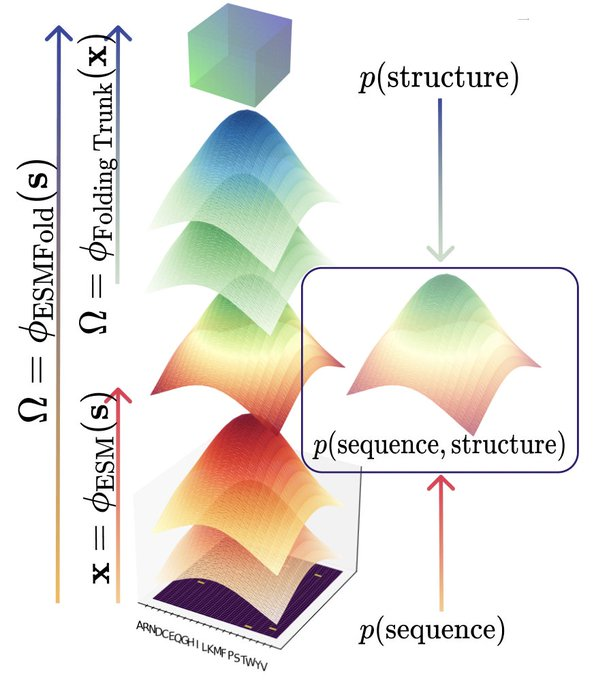
Happy to release CHEAP: Compressed Hourglass Embedding Adaptations of Proteins
We compress the ESMFold latent space while retaining information about sequence, structure, and function, with implications for generation, search, and transfer learning.
Tokenized and Continuous Embedding Compressions of Protein Sequence and Structure
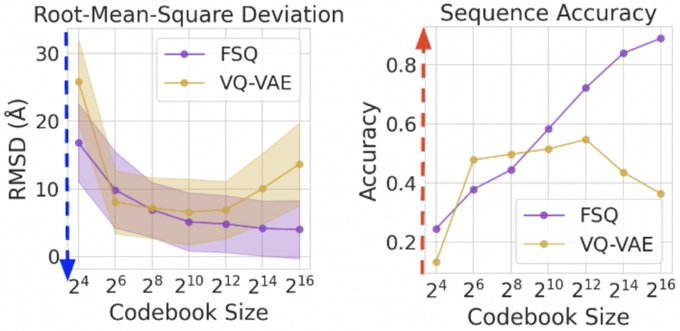
- CHEAP, a novel method for compressing protein sequence and structure latent space (ESMFold), achieves up to 128x channel and 8x length compression from sequence input alone
- Uses per-channel normalization, downsampling both the channel and length dimensions with linear projections and attention
- Explores both continuous and discrete compression, evaluated with TM-Score, RMSD, RMSPD, and sequence recovery accuracy
Preprint: https://t.co/L8hZXNjZ9u
Age-based memory decline correlates with quality—not quantity—of dendritic spines in the temporal cortex, a new @ScienceAdvances study suggests. https://t.co/6aKdcMLCkc
📢 Finally out in @J_A_C_S: We engineered the first #PhotoLanZyme, a lanthanide-dependent photoredox enzyme that catalyzes radical C-C bond cleavages upon visible-light irradiation!
💡🧫🧪🧬💻
@TU_Muenchen@ERC_Research
https://t.co/9OCFRHhzX4
Fast, sensitive detection of protein homologs using deep dense retrieval | @NatureBiotech
-Dense Homolog Retriever (DHR) employs a bi-encoder (ESM1b, first vector as fixed-length vector) architecture and a CLIP-like approach to train on homologous pairs with in-batch negatives
- Retrieve homologs by directly comparing these embeddings using a similarity metric (dot product) and JackHMMER to construct MSAs
Link: https://t.co/i8fgd2rrjX