NEW: Postdoctoral Fellow with @ewanbirney
The project is based in the Birney Research group at @emblebi and working closely with Open Targets and @MoritzGerstung's group at DKFZ
Got a PhD in bioinformatics or quantiative/CS degree? Want to work with AI on real world health care data, using some of the biggest datasets in the world? Want to live in Cambridge UK and work in European EMBL? This job in my research group is for you! https://t.co/357ZzgVZpx
New preprint 📣
In this work with @IanBrettell we developed a behavioural assay for medaka fish and the associated downstream data analysis pipeline.
https://t.co/KsF1tuSfxu
📢Our preprint on deep learning for carotid plaque detection in 170,000 UK Biobank ultrasound images is out❗️
👉robust associations of plaque presence/count with cardiovascular events🧠🫀
👉largest GWAS for carotid plaque (29,790 cases/36,847 controls)🧬
https://t.co/jVKwYaG6IB
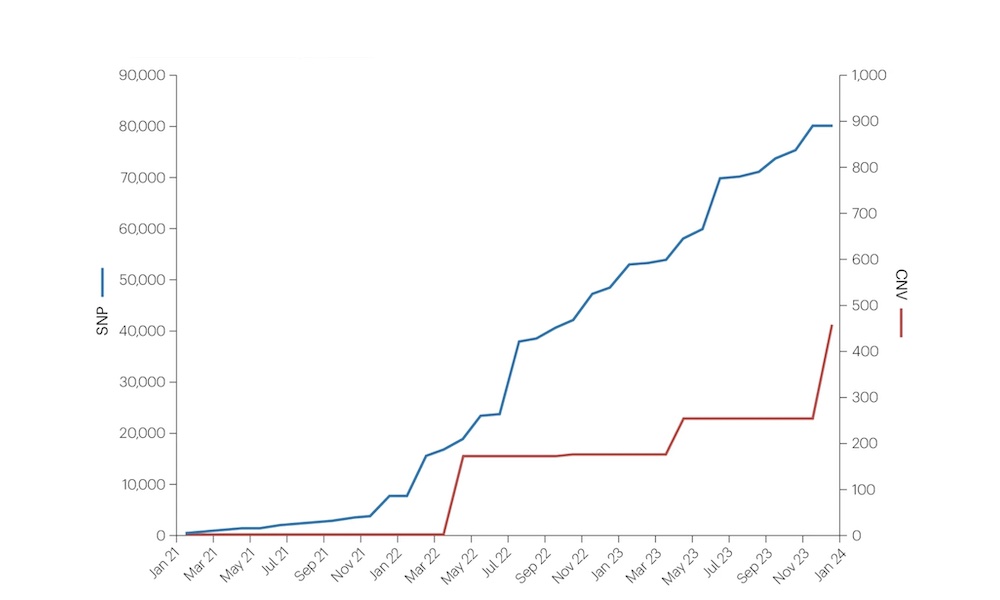
Genome-wide association studies focusing on SNPs have advanced our understanding of certain diseases but are reaching limits for some conditions.
This @NatureRevGenet review highlights how exploring copy number variation can help break these limits.
https://t.co/zQvXWC8jC6
Looking to the future, we ask what kind of genetic variation will be a priority for genome wide studies in the coming years, and how can we make sure the data is FAIR? Read more @NatureRevGenet 👇Thanks to @tomaswfitz & all involved! https://t.co/RM0RByN0gZ
This was a really fun review to write together, interesting to reflect on why it’s taken so long for CNVs to level up to similar large scale GWAS applications - lots still to do but slowly getting closer to the SNP GWAS world 😀
Got a PhD in a quantitative science? Want to work on AI and healthcare data? Want to work with one of the most innovative Pharma companies worldwide? Want to live in Cambridge UK and work across Europe? Apply below (position #2 :))
We have a new preprint out 🥳. FlexLMM is a Nextflow pipeline for running linear mixed models for GWAS that focuses on flexibility: users can specify the statistical model to be tester and the null model as R-style formulas. (continued)
https://t.co/QwKafMwLU7
Excited to share that my first first-author paper is online on @BioinfoAdv ! Check it out and thanks to amazing collaborators @WelzBettina@WittbrodtLab and supervisor @ewanbirney@tomaswfitz
See the thread for a brief explanation of the work we did
https://t.co/OycOVJtojP
A new preprint - and first from my lab from @saulpierotti - on a technical subject which we were frustrated by the lack of explicit exploration in the literature: how to best perform low coverage F2 sequencing to impute full genotypes from a F2 population. https://t.co/FPBhgi5Y4O
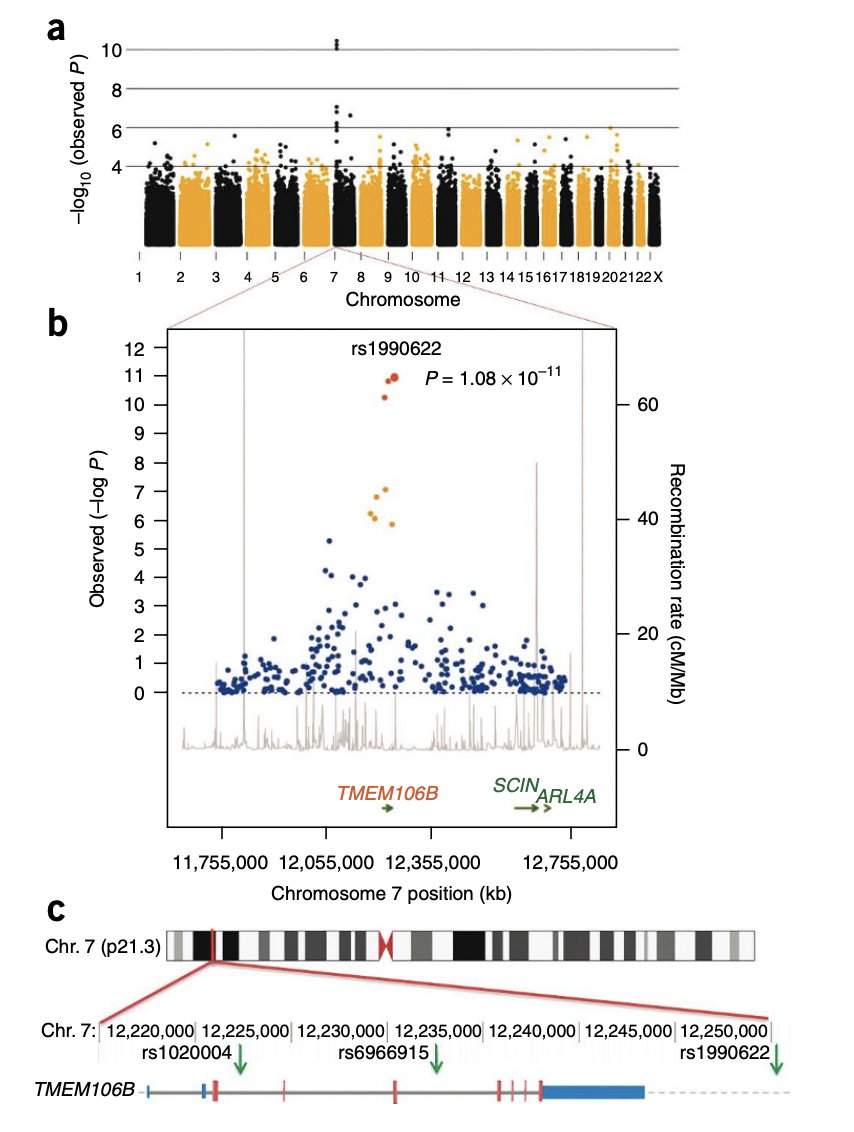
In 2010, the first genome-wide significant locus for frontotemporal lobar degeneration (FTLD) was discovered--an LD block spanning TMEM106B. Now, thirteen years later scientists found the causal variant--a 322 bp 3' UTR deletion.
When it comes to structural variants in humans, we are still scratching the surface. The situation is likely to change soon, with whole genome sequencing and long-read sequencing technologies being used more and more recently, both in research and in clinics.
Research institutes like deCODE have already given us a glimpse into what we will find once we start long-read sequencing structural variants at scale: genetic signals at many of the GWAS loci are in fact driven by large structural alterations of non-coding (and coding) regions (https://t.co/iwgoNaqwVD).
The FTLD TMEM106B locus is another beautiful addition to the list of GWAS loci driven by structural variants.
The 7p21 locus containing TMEM106B first surfaced in 2010 based on a GWAS of 515 FTLD cases and ~2500 controls (https://t.co/dIWg1zmET8). The authors found multiple correlated SNVs at this locus and had no clue which one was driving the signal. But the most interesting observation was that the minor allele of the top SNV was protective; it decreased the odds of FTLD in those carrying GRN mutations (the most frequent genetic cause of FTLD) by ~40%.
Though the causal variant isn't clear, one thing that got people excited is that one among the correlated SNPs is a TMEM106B missense variant. To test if this missense variant is what confers protection in the background of GRN haploinsufficiency, scientists knocked this variant into the mice with and without Grn. Unfortunately, they couldn't replicate the protective effect of this variant in mice. (https://t.co/w013Tx9ZzK).
Now, Chemparathy et al. from Stanford long-read sequenced the TMEM106B locus in ~400 individuals and discovered that the top risk variants at this locus are in perfect concordance with a 322 basepair deletion of the 3' UTR region, which they further replicate in an independent dataset.
UTR regions control the expression at the post-transcription levels. In agreement with that, this deletion decreased the TMEM106B protein levels but not the mRNA levels.
Though these findings do not fully establish that this structural variant is the causal one, they offer a compelling biological hypothesis that other scientists will probably soon follow up with experiments.
It always blows my mind to see how long it takes to crack open a GWAS signal, particularly the early ones, which are often described as the "low-hanging fruits".
https://t.co/X4bnl1tiHL
🐟We have a few last-minute places on offer for our free, virtual #medaka#fishmodels workshop next week!
🔗Secure your place now: https://t.co/T2tn6Edtde
🤔Already registered? Your access links will be available on the course website by 7:00 BST on Monday 17 October.
Are you a scientific researcher with experience in using #fishmodels for #genetic & #phenotypic analysis with an interest in Gene by Environment effects?
📅 17 – 18 October
👥In association with @NIEHS & @ERC_Research.
🔗Register for our free workshop: https://t.co/T2tn6EcVnG
Copy number variation (CNV) in the genome can lead to complex human diseases, including neurological disorders.
CNest is a new, open source method for robust CNV discovery from whole genome sequencing datasets.
https://t.co/eM4qA64XEu
🎺New paper published🎺
CNest, the shiny new, "do real proper /common/ CNV association studies" off Whole Exome or Whole Genome data from @tomaswfitz and myself has been published today from @CellGenomics https://t.co/GaDXPbjfs3. 2 meta points: