el ingeniero que construyó Claude Code acaba de publicar un video de 28 minutos sobre cómo escribir prompts que realmente funcionan
he visto cursos de 300$ que no cubren lo que él muestra en los primeros 10 minutos
archivos CLAUDE.md, atajos de memoria, sesiones paralelas, patrones de prompting
todo en un video y completamente gratis
funciona seas desarrollador, principiante o alguien que lleva meses usando Claude
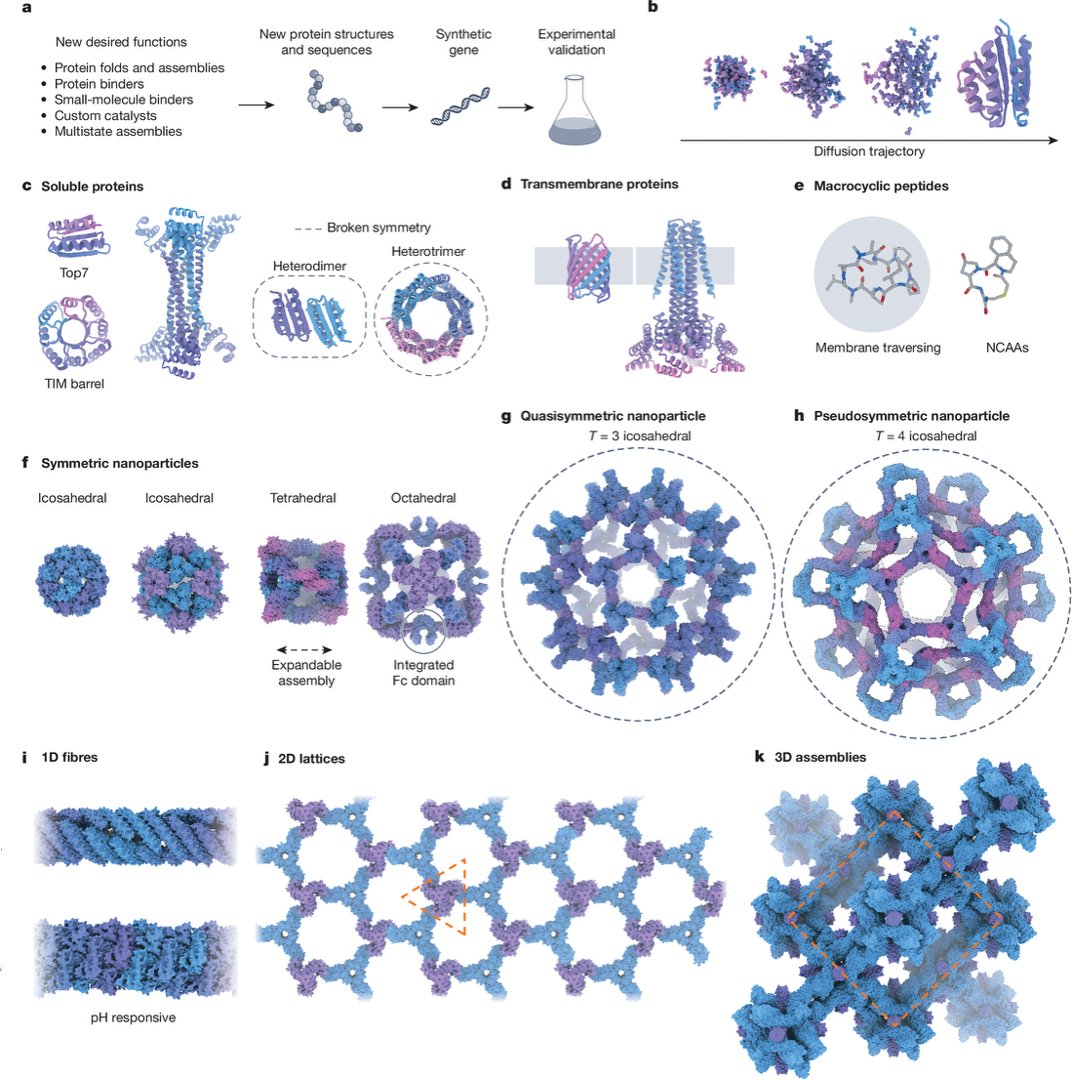
1/ Excited to share our new Review in @Nature:
“The past, present and future of de novo protein design.”
Here, we mainly focused on structure-guided protein design. The field is entering a new phase: now that we can design new proteins, what should we build next?
Predicting RNA 3D structure is a grand challenge of biology.
2 new approaches published today to get at it
https://t.co/KQ2oCGlUrN @MolecularCell
https://t.co/ofupia6du2 @NatMachIntell
Small-molecule binding and sensing with a designed protein family @NatureComms
🚀 New paper from David Baker!🚀
1 Researchers have developed a powerful new computational strategy that combines deep learning with physics-based methods to design a family of proteins capable of binding diverse small molecules with high affinity and atomic-level precision.
2 The core innovation lies in the diversification of the NTF2-like fold using structure generation algorithms to create over 10,000 unique scaffolds with varied internal pocket geometries tailored for ligand docking.
3 This approach successfully produced functional binders for six chemically distinct targets, including hormones like cortisol and drugs such as the anticoagulant apixaban, with binding affinities reaching the nanomolar range.
4 The design process utilized LigandMPNN, a deep learning model trained specifically on protein-ligand complexes, which proved highly effective at generating accurate protein-ligand interactions that were later confirmed by crystal structures.
5 Beyond simple binding, the study demonstrates a modular platform for sensor development by creating a cortisol-induced heterodimerization system that functions as a bioluminescent biosensor at physiologically relevant concentrations.
6 High-resolution structural analysis revealed that the designed proteins match their computational models with sub-angstrom accuracy, validating the reliability of these new deep learning-integrated design pipelines.
7 This work establishes a versatile foundation for designing custom sensors and binders for a wide array of applications across environmental monitoring, diagnostics, and therapeutics.
📜Paper: https://t.co/xCmW8yDkzs
#ProteinDesign #DeepLearning #Biosensors #ComputationalBiology #Biochemistry #NatureCommunications
The therapeutic potential of orphan adhesion G-protein-coupled receptors
https://t.co/S3VGtldvWY
https://t.co/DYmMOQTthf
This Review discusses opportunities to target aGPCRs in cancer, psychiatric disorders and autoimmune diseases, based on advances in structural biology
Co-folding of Membrane Proteins and Lipid Molecules Improves Membrane-Protein Structure Prediction Accuracy
1 This study introduces CoMPLip, a straightforward protocol that feeds lipid molecules into AlphaFold 3 as co‑folded inputs, allowing the model to predict a protein and surrounding lipids simultaneously.
2 During prediction, the lipids self‑assemble into bilayer‑like arrangements around transmembrane helices, thereby providing an explicit membrane environment that was previously only implicitly encoded in training data.
3 In a detailed case study of the E. coli protease RseP bound to batimastat, adding 100 molecules of 1‑monoolein reduced the ligand RMSD from 10.79 Å (no lipids) to 1.37 Å, while the protein backbone remained similarly accurate.
4 A benchmark of 65 experimentally determined membrane‑protein–ligand complexes showed that CoMPLip produced correct ligand poses for four additional targets, raising the number of correct predictions from 41 to 45, though the overall success rate remained unchanged.
5 For 123 full‑length single‑pass proteins, CoMPLip enabled clear extracellular–intracellular domain separation in 61 cases versus only 20 without lipids, demonstrating that the lipid layer physically blocks domain contacts across the membrane.
6 When applied to the dynamic transporter NTCP, CoMPLip sampled both inward‑facing and outward‑facing conformations (approximately 60 % each), whereas standard AlphaFold 3 predicted only the inward state, illustrating enhanced conformational diversity.
7 To mitigate the influence of the many added lipids on the ranking score, the authors devised SCoMPLip, a protein‑ligand‑specific metric that weights protein and ligand pTM scores and the protein–ligand interface, improving model selection for the target complex.
8 Practical guidelines suggest using about 100 copies of 1‑monoolein for a four‑pass transmembrane protein; however, this increases GPU memory demands and may limit applicability to very large targets.
9 The authors note that CoMPLip’s benefits are modest for ligand pose accuracy overall, and that lipid placement can sometimes be unphysical (e.g., occupying the pore); future work will explore other co‑input molecules and broader ranking‑score optimization.
10 Overall, CoMPLip offers a training‑free, easily implementable enhancement for AlphaFold‑based membrane protein modeling, adding an explicit membrane context that improves ligand binding, domain assembly, and state sampling in realistic environments.
📜Paper: https://t.co/N2ZmsYkln4
#ProteinStructure #AlphaFold3 #MembraneProteins #ComputationalBiology #Bioinformatics #StructuralBioinformatics
Quantum computing just touched real biology.
For the first time, scientists at IBM and Cleveland Clinic simulated the electronic structure of a real protein 👀
They used a hybrid system called quantum centric supercomputing (QCSC) to model the 303-atom Trp-cage miniprotein.
Instead of forcing a quantum computer to do everything, they split the problem intelligently:
Classical computers handled the easier regions, while quantum processors tackled the hardest part, strong electron correlations.
Using wavefunction-based embedding (EWF), the protein was broken into smaller quantum-relevant clusters and solved efficiently.
And NOTE, this is NOT protein folding like AlphaFold.
This goes deeper.
It simulates how electrons behave inside molecules, the level that actually determines chemical reactions, drug binding and molecular stability.
Right now, drug discovery still relies on approximations and trial and error.
But if we can simulate molecules at the true quantum level, we move toward designing drugs before lab testing, predicting reactions with high accuracy and even creating entirely new materials.
Large Scale Prospective Evaluation of Co‑Folding Across 557 Mac1‑Ligand Complexes and Three Virtual Screens
1. The study prospectively tests AlphaFold3, Chai‑1, and Boltz‑2 on 557 newly crystallized SARS‑CoV‑2 Mac1 inhibitors, achieving >50 % of ligand poses within 2 Å RMSD—outperforming the classic DOCK3.7 benchmark.
2. Pose accuracy remains high even when the test ligands share little structural similarity (low ECFP4 Tanimoto or MCS%) with any ligand in the models’ training sets, indicating genuine generalization rather than memorization.
3. Co‑folding confidence scores (AF3 L‑pLDDT, Chai‑1 ipTM) discriminate accurate poses with AUCs of 0.76 and 0.73, while Boltz‑2’s predicted pIC₅₀ correlates strongly with experimental potency (r = 0.60) and yields MAEs ≈0.7 kcal after linear calibration.
4. When applied to large‑library hit lists (AmpC β‑lactamase, σ₂ receptor, dopamine D4), co‑folding did not surpass docking in separating true binders from false positives; DOCK3.7 maintained higher enrichment and AUCs.
5. The complementary strengths suggest a workflow: use docking for rapid hit identification, then apply co‑folding for accurate pose prediction and affinity ranking within a discovered series to guide optimization.
6. This is the first extensive prospective assessment of co‑folding on a diverse, industrial‑scale dataset, demonstrating that deep‑learning models can reliably inform structure‑based drug design beyond traditional docking.
💻Code: https://t.co/DQK0hnPXI0
📜Paper: https://t.co/fzH9yWYpzp
#DeepLearning #ProteinLigand #StructureBasedDrugDiscovery #CoFolding #AlphaFold3 #Boltz2 #Chai1
Our paper on inward H+ pumping rhodopsin, Schizorhodopsin (SzR), from Antarctic bacteria—by former member Dr. Marín, Prof. Béjà and Dr. Rozenberg (Technion)—is now published in Biophys. J.!🎊🦠SzRs are conserved across a broad range of species 📄
https://t.co/x262HBOzIr
@BiophysJ
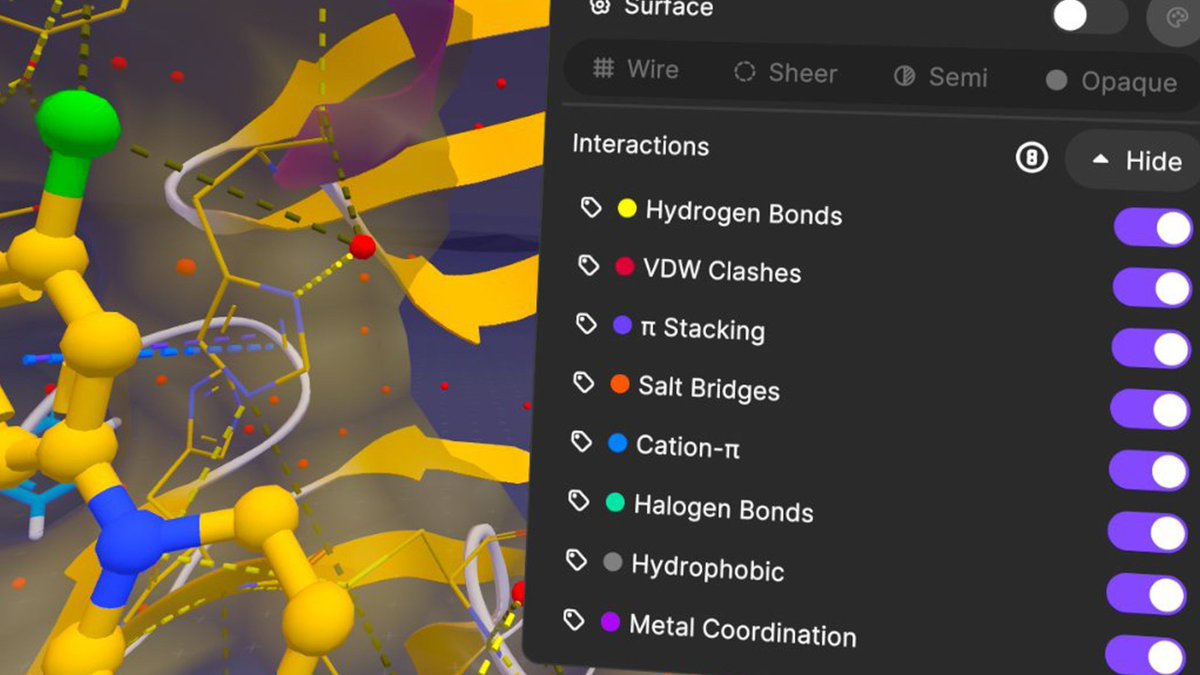
See every chemical interaction in XR
Nanome v2.4 introduces native chemical interaction calculations.
H-bonds. Salt bridges. π-stacking. Halogen bonds. Hydrophobic. Metal coordination. VDW clashes.
Move your ligand and watch interactions update instantly.
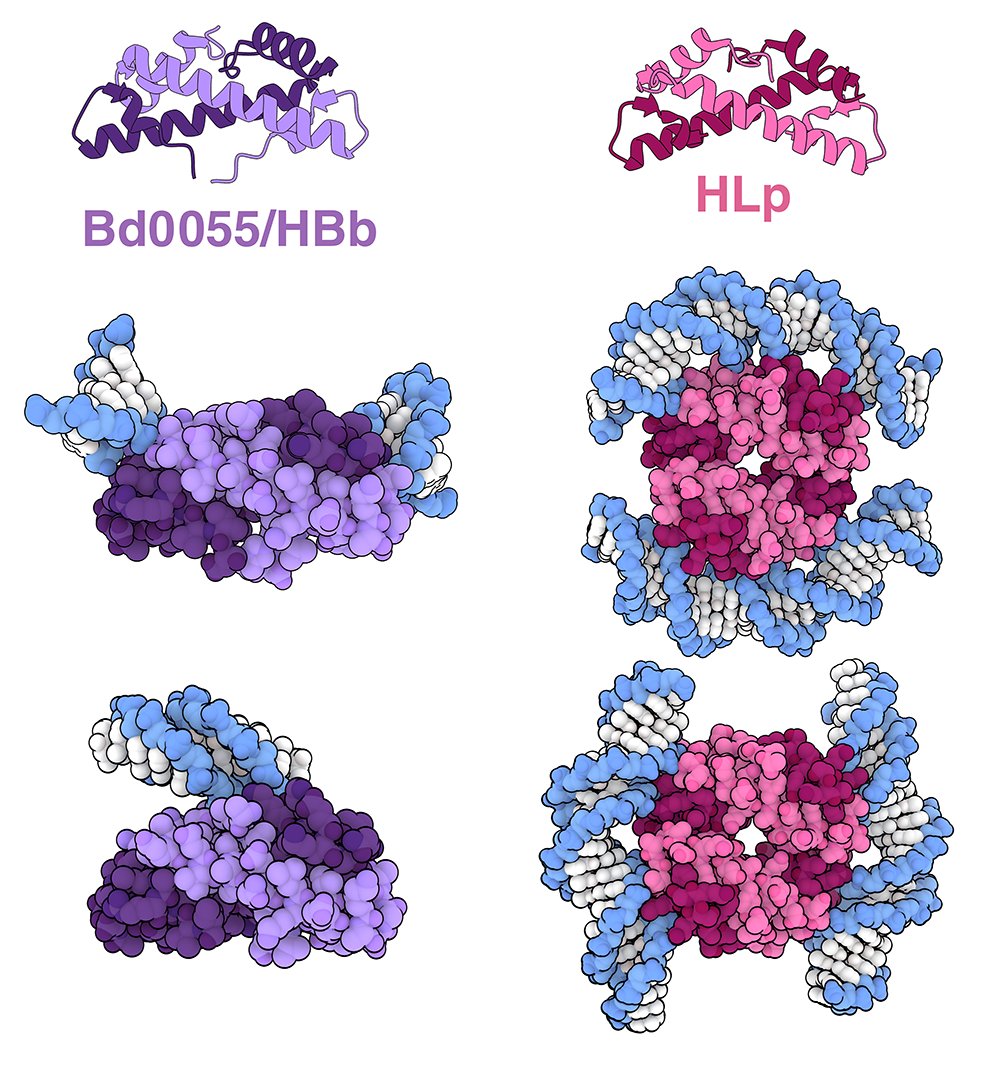
Surprisingly, histones have also been characterized in bacteria, including a bacterium found in soil and aquatic environments and in a spiral-shaped gram-negative bacterium
More at Molecule of the Month: https://t.co/AuVysaeXWw