My PhD work preprint is now online.
We identify a lipid-dependent allosteric site that determines how LC3 interacts with its binding partners at the autophagosome membrane
Read here: https://t.co/2Mx7z864ZI
@jesucastin@Sci_Lipi@IGIBSocial
Happy to share our recent work that took a long time in making!
What do lipids DO to PROTEINS? Well, we show that lipids allosterically open the receptor binding pocket of LC3, a key protein in autophagy. Using this fundamental mechanism, we engineered LC3 proteins that are either functionally “inactivated”or “hyper-active”on autophagosomal membranes
A novel lipid-triggered allosteric site modulates LC3-LIR receptor binding activity https://t.co/PmLJGCSU7k
A consortium of 20 research institutions across the country has come together to develop a DNA map of the nation. Read about this exciting research "The GenomeIndia Project" here:
https://t.co/boszPQFhjV
@CSIR_IND@DBTIndia@souvik_csir@IGIBSocial
Congratulations to GalaxEye on the successful launch of Mission Drishti!
A significant milestone in India’s space journey, with
world’s first OptoSAR satellite and India's largest privately built satellite, advancing all-weather Earth observation capabilities.
ISRO is proud to support this achievement by enabling access to its satellite testing facilities fostering innovation and handholding India’s emerging private space sector.
Best wishes to the team at @GalaxEye for continued success in strengthening the nation’s space ecosystem and advancing cutting-edge technology.
We don't even design binders!!
But we @BioMandrake just won @adaptyvbio × @gembioworkshop's RBX1 design competition - 1 Strong binder out of 322 tested, selected from 12,000+ submissions.
Couldn't make it to @iclr_conf in Rio. Here's how we did it 👇
https://t.co/Vt5lzgDRNB
For decades, biology textbooks have enshrined a simple rule: DNA is made by copying a template. After one enzyme unzips a DNA double helix into separate strands, another called a polymerase builds a complementary sequence, base by base, for each strand. Presto: two copies of the original DNA.
But new research into how bacteria defend themselves from viruses now shows this synthesis rule isn’t absolute.
Now, a team describes a bacterial enzyme that synthesizes DNA without a nucleic acid template, using its own structure as a guide.
Learn more: https://t.co/bpVgr0KMdR
We made Caliby much easier to use! Try it out:
Colab notebook: https://t.co/4uwRnRV4Px
HuggingFace Spaces: https://t.co/3AOP0i5uTw
pip install it and use the Python API: https://t.co/dQlFM9eLGS
the blogs that @tamarindbio write legitimately make it possible to understand the whole landscape from one article
every other field needs this, and no other field has it
What if AI could invent enzymes that nature hasn’t seen? 👩🔬🧑🔬
Introducing 🪩 DISCO: Diffusion for Sequence-structure CO-design
14 rounds of directed evolution and over a year of wet lab work. That's what it took to engineer an enzyme for selective C(sp³)–H insertion, one of the most challenging transformations in organic chemistry.
DISCO surpasses this with a single plate. No pre-specified catalytic residues, no template, no theozyme, no inverse folding, just joint diffusion over protein sequence and structure.
📝 Blog: https://t.co/j9Za0JigfO
📄 Paper: https://t.co/ficrYNBBrM
💻 Code: https://t.co/p81sSwoaPH
A new best-in-class structure predictor AND de novo design protocol
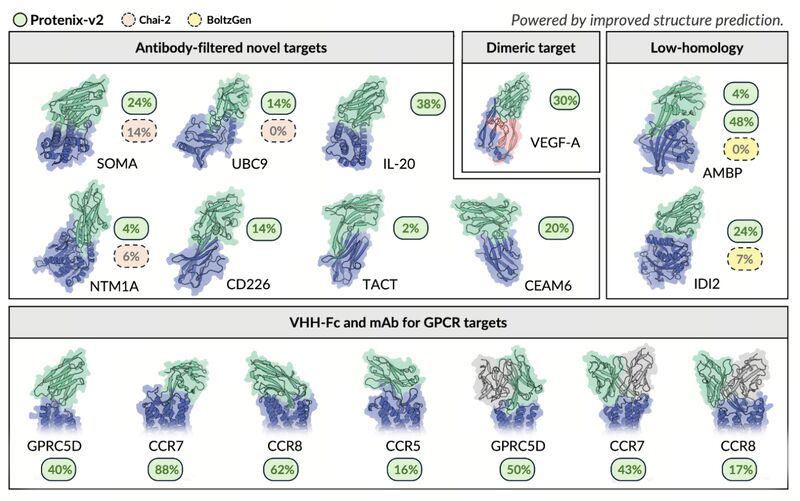
Protenix-v2 claims to outperform AlphaFold3 in antibody-antigen structure prediction tasks, showing a 13% increase over its previous generation in DockQ scores.
Available on @tamarindbio today.
Protenix-v2 with only 5 seeds beats Protenix-v1 with 1000 seeds on antibody–antigen prediction. This implies a technical improvement, while not needing to massively scale inference of a given model like other providers previously showed.
In addition, the authors use Protenix-v2 as a scoring and ranking mechanism for de novo antibody design. They report a 100% target-level success rate on the current soluble-target panel, meaning at least one confirmed binder for every tested target, with BLI-confirmed VHH-Fc hit rates from 2% to 48%. They also show that epitope choice matters a lot: on AMBP, one epitope gave 4% hit rate and another 48%.
The GPCR result is probably the most impressive experimental result in the paper. With only 16–30 tested designs per target, the protocol shows VHH-Fc hit rates of 16%, 62%, 40%, and 88% across four GPCRs, and corresponding mAb hit rates of 0%, 17%, 50%, and 44%. They also report a best GPRC5D VHH-Fc binder of 112 pM under avidity conditions.
Congratulations to the @ai4s_protenix team on the release!
Molecular design for AI agents: announcing the Tamarind MCP Server.
Today, scientists can use the @tamarindbio library of 250+ molecular design tools(Boltz, AlphaFold, RFdiffusion...) in any AI chat interface.
We serve not just open-source models, but the internal protocols your team has onboarded to Tamarind. Any tool added to Tamarind is then available across the MCP server, Tamarind web app, and API, so it can be used in chat-based agents, multi-step pipelines, ELNs, and LIMS-connected workflows.
Our goal is simple: make Tamarind the place where scientists can access the BioAI tools they need, wherever they want to work, while we handle the infrastructure.
Many users have already incorporated our MCP into their internal AI agents, along with community efforts like Blatant-Why building apps on top of Tamarind. Try out our tooling for antibody design, small molecule virtual screening, developability/ADMET scoring and more!
Extremely excited to announce LigandForge 🧬⚡
Generate high-quality peptides at over 10,000x - 1M the speed of state-of-the-art methods like Bindcraft and Boltzgen. Predict binding affinity with 83% correlation to experimental binding data. 150 protein targets benchmarked.
At @GoogleDeepMind, we believe AI is the ultimate catalyst for science. 🧬
The best example of this has been the AlphaFold database (AFDB) of protein structure predictions which has been used free of cost by more than 3.3 millions researchers across the world!
Today, in collaboration with @emblebi, @Nvidia and @SeoulNatlUni, we are expanding the database by adding millions of AI-predicted protein complex structures to the AlphaFold Database. To maximise global health impact, we’ve prioritised proteins that are important for understanding human health and disease, including homodimers from 20 of the most studied organisms, including humans, as well as the @WHO’S bacterial priority pathogens list.
Read more here:
https://t.co/RZGq8vrIPS
Learning the All-Atom Equilibrium Distribution of Biomolecular Interactions at Scale
1 ByteDance AI Drug Discovery and Anew Therapeutics researchers introduce AnewSampling, the first generative foundation framework that faithfully reproduces molecular dynamics (MD) at the all-atom level for sampling the equilibrium distribution of biomolecular interactions, addressing the high computational cost of traditional MD simulations.
2 AnewSampling leverages a novel quotient-space generative framework to ensure mathematical consistency in its modeling, and is trained on AnewSamplingDB—the largest self-curated database of protein-ligand trajectories to date, containing over 15 million conformations across 10,297 unique protein sequences and 27,979 unique ligand SMILES.
3 The framework builds on an AlphaFold3-like architecture with a stratified hybrid fine-tuning strategy: Low-Rank Adaptation (LoRA) for sequence representation modules and full-parameter fine-tuning for the Diffusion Module, alongside a Cluster-Based Template Guidance mechanism to enforce exhaustive exploration of the equilibrium ensemble.
4 In benchmarking on the ATLAS monomer dataset, AnewSampling outperforms all state-of-the-art generative methods across all 13 evaluation metrics, showing unparalleled accuracy in predicting protein flexibility and distributional accuracy for monomeric systems.
5 For protein-ligand dynamics testing across held-out PDB systems, JACS & Merck industrial datasets and an in-house drug discovery pipeline dataset, AnewSampling achieves statistical alignment with ground-truth MD distributions that far surpasses static predictors and MD-enhanced models like Boltz2, with its generated conformations nearly indistinguishable from MD baselines in key metrics.
6 AnewSampling demonstrates emergent enhanced sampling capabilities beyond conventional MD, successfully navigating high energy barriers to recover coupled ligand and side-chain motions in CDK2 systems (1H1R and 1H1S)—a major challenge for traditional MD that often requires replica-exchange MD (REMD) to achieve.
7 The model accurately captures subtle ligand-induced conformational shifts in congeneric structure-activity relationship (SAR) series, a critical capability for lead optimization in drug discovery, and maintains high fidelity in modeling non-covalent protein-ligand interactions and global protein backbone dynamics across diverse chemical and conformational spaces.
8 The research team proposes a multi-level assessment strategy for generative biomolecular dynamics models, using metrics like Jensen-Shannon (JS) distance for ligand torsion, Wasserstein (WS) distance for protein-ligand interactions and Spearman correlation for Cα RMSF to rigorously validate physical fidelity at the atomic level.
9 AnewSampling offers unprecedented computational efficiency for exploring biomolecular conformational landscapes, enabling integration into research and industrial drug discovery pipelines and driving a shift toward dynamics-aware design of adaptive inhibitors and functional biomolecules.
10 While AnewSampling achieves significant advances, the researchers note current limitations including reliance on structural templates, limited training data for broader biomolecular interaction types (e.g., protein-nucleic acid) and restriction to fixed thermodynamic environments, outlining future work to address these and enable sequence-only equilibrium distribution prediction.
11 AnewSampling and conventional MD are shown to be complementary: MD provides the critical training data for the generative model, while AnewSampling can accelerate MD by generating diverse initial structural candidates that help bypass energy barriers in physical simulations.
📜Paper: https://t.co/bBaP2DPtvu
#AIDrugDiscovery #BiomolecularDynamics #AllAtomModeling #GenerativeAI #ComputationalBiology #MolecularDynamics #ProteinLigandInteractions
Scientists say they have made some of the first direct measurements of how long it takes an individual, ordinary protein to fold – and the results were surprising.
https://t.co/gs7UUNBLke
How to Design Antibodies (with AI)
https://t.co/lkybBrPGm6
@btnaughton is one of the best applied practitioners in the rapidly evolving world of computational protein design.
Today, he published a step-by-step primer on the five steps that go into this new science:
1. Choosing a Target
2. Preparing the Target Structure
3. Running a Design Campaign
4. Filtering and Selecting Candidates
5. Experimentally Validating the Results
Worth reading to better understand this frontier. It's also a great resource for understanding how the different models and products in this space can be compared right now.