Our new manuscript for DeepArk is now online @genomeresearch ! We developed #DeepLearning models of the regulatory code for mice, C. elegans, Drosophila, and zebrafish! https://t.co/plCjL8qaeM
#Genomics#MachineLearning
I've raised about $100 million for biotech projects at this point - fundraising can be really hard.
I wrote up thoughts for newer biotech founders trying to raise in the new year (longer post at end) 🧵
Come for the fun name, stay for the interesting benchmarks!!
the language of non-coding DNA is v mysterious and sort of wild west 🤠🐄 compared to the land of proteins --> we start to crack that with a new model LOL-EVE! neat work with @c_sheare and team!! : )
We made sequences generated by EvoDiff in the lab!
- Unconditional generations express, fold, and exhibit expected secondary structure elements
- We inpaint functional intrinsically-disordered regions and scaffold binders
If you're looking for labs working on Protein Design, we've put together a list based on our best knowledge. The initial list came from @Zuricho_zbzt, and with a bit of help from me, we've now got it up on GitHub. Feel free to suggest any labs we might have missed!
https://t.co/21YP6rQGJV
It is *incredibly* easy to game the LLM benchmarks. Training on test set is for the rookies. Here're some tricks to practice magic at home:
1. Train on paraphrased examples of the test set. "LLM-decontaminator" paper from LMSys found that you can beat GPT-4 with a 13B model (!!) on MMLU, GSK-8K, and HumanEval (coding) just by rewriting the exact same test question in different formats, phrasing, or even foreign languages. Easily +10 point gains.
2. It's easy to game the LLM-decontaminator as well. It only checks for paraphrasing, but you can use any frontier model to generate *new questions* that are different on the surface but very similar in solution template/logic. In other words, you attempt to overfit to a close distribution of the test set, but not to individual samples. HumanEval, for example, is a bunch of simple Python questions (i.e. a specific, narrow distribution) that do not reflect the real world coding complexity at all.
3. You can also prompt-engineer the heck out of your generator to fool the LLM-decontaminator or whatever detector. The detector is public but your data gen is private. Take advantage of that.
4. Increasing inference-time compute budget almost always helps. Self-reflection is a technique known for a long time (see Reflexion, Shinn et al. 2023). Also try simple majority voting or Tree of Thought. These thought traces are essentially test-time ensemble methods, and the more the merrier. It's obvious that ensemble of N things > 1 thing if you don't control for inference-time tokens.
It's incredible that people still get excited by MMLU or HumanEval numbers in Sept, 2024. These benchmarks are seriously broken, and gaming them can be an undergrad homework project.
I would not trust any claims of a superior model until I see the following:
1. ELO points on LMSys Chatbot Arena. It's difficult to game democracy in the wild.
2. Private LLM evaluation from a trusted 3rd party, such as Scale AI's benchmark. The test set must be well-curated and held secret, otherwise it quickly loses potency.
1/ I did my own little hackathon last weekend designing EGFR binders for @adaptyvbio's protein design competition. I was really excited to see that my submissions took the top 10 spots in the virtual scoring phase! I got some DMs asking about my process so here's a thread:
Title:
AlphaFold3, a secret sauce... 😇
Acknowledgments and Disclosure of Funding:
We thank our colleagues for submitting the test queries to the AlphaFold3 server...
Introducing Genie 2, our latest protein design model! Genie 2 sets a new state of the art on key design metrics and supports motif scaffolding, including that of multiple motifs with undefined geometric relationships, a feature new to protein diffusion models. (1/5)
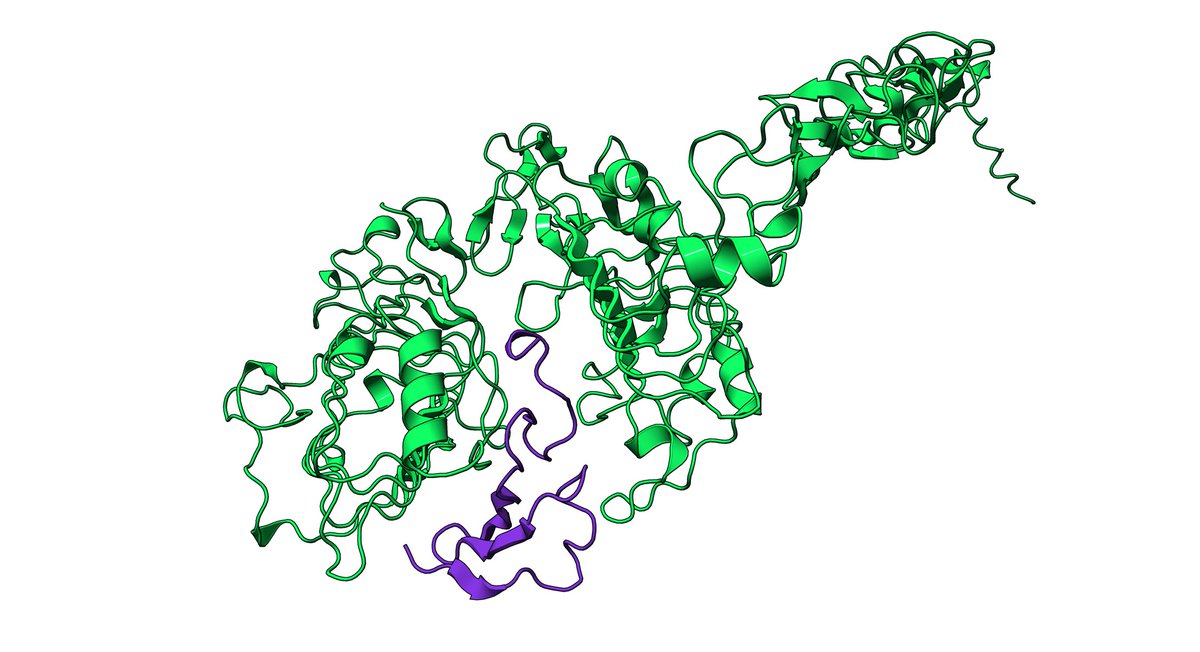
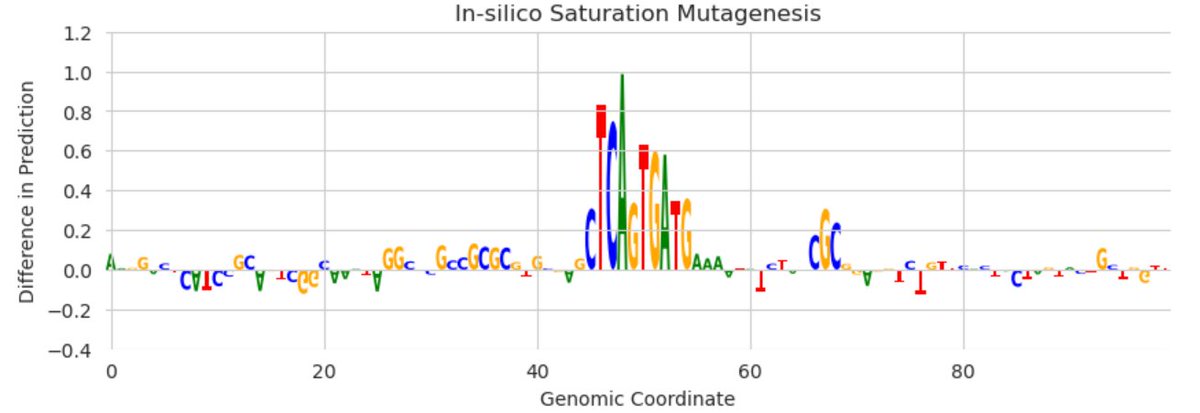
You've probably seen attribution tracks where the height of each letter is its "importance" to a predictive model and motifs pop out.
But the technical details behind how these are calculated can matter a lot -- and I'm worried many may be done incorrectly. 1/
A Mamba Primer (w/ Yair Schiff https://t.co/GnYleKalcW )
Mamba is a nice jumping off point to summarize foundational ideas in sequence modeling, parallel algorithms, continuous-time representations, and GPU aware algorithms. We try to put these together in the context of LMs.
It’s a story of how academia goes wrong. And how it self-corrects.
A must read for everyone:
2020 - A discovery of room-temperature superconductivity is published in Nature by Ranga Dias
2023 - The second RT superconductor is reported by Dias in Nature
2024 - The first paper is retracted. Then the second. No one could replicate his results.
The forth investigation by external experts showed that ‘he concealed information from his students, manipulated them and shut them out of key steps in the research process’.
▫️
The story looks like this:
1. Dias proposed that adding carbon to H3S might lead to RT superconductivity.
2. His PhD students could NOT observe the signs of superconductivity in the samples (no resistivity drop, no susceptibility change).
3. Suddenly, Dias emailed everyone and announced the discovery of RT superconductivity. Students were shocked.
4. The draft was sent to the team at 5.13 p.m. and SUBMITTED to Nature at 8.26 p.m. In just TWO hours! No one had time to read it.
5. Dias told the students he ‘had taken all the resistance and magnetic-susceptibility data before coming to Rochester’.
6. 3 reviewers were concerned. Only one supported publication. It was published on 14 October 2020.
7. A year(!!!) later Dias made raw data publicly available. This prompted data check where ‘data points were separated by suspiciously regular intervals’.
8. Then, Nature initiated post-publication review by 4 experts. Two said there were big problems.
9. Following rebuttal & responses, Nature retracted the manuscript. Dias did NOT tell the students about the post-publication review process and data fabrication concerns.
▫️
Now, the 2nd paper raised concerns about PhD students:
- Dias sent them the draft at 2.09 am and asked for comments by 10.30 am (same day). The draft had NO figures.
- The students convinced Dias to postpone submission by one day.
- The students raised concerns about the pressure data because none of it was “anything that we actually measured”. Dias dismissed it with: “Pressure is a joke.”
- Then ‘Dias gave them an ultimatum: remove their names, or let him send the draft’. It was very intimidating. The students gave in: “What if I [object] and he makes the rest of my life miserable?”
▫️
My conclusions:
1. This situation shows how easily a supervisor can manipulate inexperienced students into publishing and believing the results. I am sure there are many other examples in academia that just haven’t surfaced.
2. All data must be publicly available. Before publication.
3. Nature took the case professionally, asking for extra refereeing and post-publication review multiple times as things were unfolding. BUT it's not so common. I heard many stories where some journals just didn’t care.
4. Finally and MOST importantly - students suffer the most.
As one of them said:
“My thesis is going to be full of fabricated data. How am I supposed to graduate in this lab?”
#academia #PhD #research
I used to find writing CUDA code rather terrifying. But then I discovered a couple of tricks that actually make it quite accessible.
In this video I introduce CUDA in a way that will be accessible to Python programmers, and I even show how to do it all in @GoogleColab!
'Universities, generally helped by their nonprofit status, have a nearly limitless, low-paid supply of research assistants to help scientists with early-stage research'
poignant !
https://t.co/G8TQvEBVmU
A few thoughts on the recent set of papers torture testing genomic deep learning for predicting individual-level gene expression [ https://t.co/rRUBH7sW6m , https://t.co/ZLcoXGPbWD , https://t.co/Je9pFg4kX2 ]. First a brief summary 🧵:
https://t.co/yUKnoeeA4r
1/n I'm thrilled to share *Deep generative modelling of the human proteome reveals over a hundred novel genes involved in rare genetic disorders* https://t.co/1xggu200pp from a wonderful collaboration between the Marks Lab and Dias and Frazer group, with @roseorenbuch as lead!🧵:





































![Andrew_Akbashev's tweet photo. It’s a story of how academia goes wrong. And how it self-corrects.
A must read for everyone:
2020 - A discovery of room-temperature superconductivity is published in Nature by Ranga Dias
2023 - The second RT superconductor is reported by Dias in Nature
2024 - The first paper is retracted. Then the second. No one could replicate his results.
The forth investigation by external experts showed that ‘he concealed information from his students, manipulated them and shut them out of key steps in the research process’.
▫️
The story looks like this:
1. Dias proposed that adding carbon to H3S might lead to RT superconductivity.
2. His PhD students could NOT observe the signs of superconductivity in the samples (no resistivity drop, no susceptibility change).
3. Suddenly, Dias emailed everyone and announced the discovery of RT superconductivity. Students were shocked.
4. The draft was sent to the team at 5.13 p.m. and SUBMITTED to Nature at 8.26 p.m. In just TWO hours! No one had time to read it.
5. Dias told the students he ‘had taken all the resistance and magnetic-susceptibility data before coming to Rochester’.
6. 3 reviewers were concerned. Only one supported publication. It was published on 14 October 2020.
7. A year(!!!) later Dias made raw data publicly available. This prompted data check where ‘data points were separated by suspiciously regular intervals’.
8. Then, Nature initiated post-publication review by 4 experts. Two said there were big problems.
9. Following rebuttal & responses, Nature retracted the manuscript. Dias did NOT tell the students about the post-publication review process and data fabrication concerns.
▫️
Now, the 2nd paper raised concerns about PhD students:
- Dias sent them the draft at 2.09 am and asked for comments by 10.30 am (same day). The draft had NO figures.
- The students convinced Dias to postpone submission by one day.
- The students raised concerns about the pressure data because none of it was “anything that we actually measured”. Dias dismissed it with: “Pressure is a joke.”
- Then ‘Dias gave them an ultimatum: remove their names, or let him send the draft’. It was very intimidating. The students gave in: “What if I [object] and he makes the rest of my life miserable?”
▫️
My conclusions:
1. This situation shows how easily a supervisor can manipulate inexperienced students into publishing and believing the results. I am sure there are many other examples in academia that just haven’t surfaced.
2. All data must be publicly available. Before publication.
3. Nature took the case professionally, asking for extra refereeing and post-publication review multiple times as things were unfolding. BUT it's not so common. I heard many stories where some journals just didn’t care.
4. Finally and MOST importantly - students suffer the most.
As one of them said:
“My thesis is going to be full of fabricated data. How am I supposed to graduate in this lab?”
#academia #PhD #research](https://pbs.twimg.com/media/GIY8mFKXgAMu1k8.jpg)




















